
Ryanodine receptors as pharmacological targets for heart disease1
Introduction
Fluctuations in cytosolic calcium (Ca2+) concentrations act to modulate a vast array of physiological processes, including neurotransmitter release, cell division, and muscle contraction. This control mechanism requires low cytosolic Ca2+ levels under resting conditions alternating with transient increases in Ca2+ upon activation. In cardiac muscle, transverse tubular invaginations of the plasma membrane contact the membrane of the sarcoplasmic reticulum (SR) Ca2+ stores to form dyadic junctions, establishing a structural framework for the external regulation of intracellular Ca2+ release[1]. Two principal classes of intracellular ion channels have evolved to facilitate the movement of Ca2+ into the cytosol from intracellular stores: (i) inositol 1,4,5-trisphos-phate receptors; and (ii) ryanodine receptors (RyR)[2]. This review article will focus on cardiac (type 2) RyR (RyR2), as increasing evidence emerges that RyR play a pivotal role in the development of cardiac arrhythmias and heart failure. We will discuss classic and novel pharmacological agents that may modulate these calcium release channels in order to treat cardiac disease.
Ryanodine receptors (RyR)
RyR consist of large tetramers of RyR monomers, comprised of a large regulatory domain protruding into the cytosol and a much smaller transmembrane domain containing the channel pore[3]. It is now well accepted that RyR2 channels exist as large macromolecular complexes comprised of numerous regulatory subunits, including calmodulin (CaM), the FK506-binding protein FKBP12.6 (also known as calstabin2), protein kinase A (PKA), CaM-dependent kinase II, protein phosphatases 1 and 2A, phosphodiesterase, junctin, triadin, and calsequestrin[4]. The gating behavior of RyR can be regulated by many of these accessory proteins (CaM, FKBP12.6, and PKA) as well as a variety of endogenous ligands (Ca2+, ATP, Mg2+)[5]. The physical and functional association of RyR2 channels results in coordinated gating behavior termed ”coupled gating”[6]. Coupled gating requires FKBP12.6 in the RyR2 macromolecular complex, and allows clusters of channels to function as Ca2+ release units that release calcium amounts that can be visualized as Ca2+ sparks[7]. As the functional effects of the RyR2 accessory subunits have been reviewed previously, this will not be the focus of the present article[4,8].
The dysfunction of RyR2 has been implicated in various diseases of the heart. A number of inherited mutations in the RyR2 gene have been identified in patients with exercise-induced ventricular arrhythmias and sudden cardiac death[9–11]. Although initially most mutations were identified in the amino terminus, central domain, and carboxy terminus, more recent genetic data suggest that mutations may occur throughout the channel protein[12]. In addition to these relatively rare genetic arrhythmias, acquired RyR2 defects have been implicated in the development of congestive heart failure[13,14]. Clinical and experimental data suggest that in failing hearts, the phosphorylation status of RyR2 is altered due to chronic hyperactivity of PKA[4,8,15]. The hyperphosphorylation of RyR2 by PKA is associated with the loss of the channel-stabilizing subunit FKBP12.6, which alters the activation properties of RyR2 and increases the open probability[13,16]. Moreover, it has been suggested that coupled gating between RyR2 may be altered in the failing heart, which might decrease systolic Ca2+ transients and/or cause a diastolic Ca2+ leak[4,6]. The unwanted diastolic leak of Ca2+ from the SR promotes the generation of arrhythmias and anomalous contraction of the heart. Although some of the mechanistic aspects of this concept have been debated in recent articles, most authors agree that a diastolic SR Ca2+ leak promotes arrhythmias and heart failure[17,18]. Therefore, RyR have emerged as novel therapeutic targets for the treatment of inherited arrhythmias and heart failure[19].
RyR pharmacology
The activity of RyR is regulated by multiple endogenous proteins residing in the macromolecular channel complex as indicated earlier. The focus of this review, however, will be on exogenous pharmacological agents that have been shown to interact with and modulate cardiac RyR[20]. These agents may be classified according to their effect on the SR Ca2+ release function (eg agonist or antagonist), or according to the mechanism of RyR2 modulation (Tables 1, 2). One class of agents modulates RyR2 primarily by altering gating of the channel (ie the opening and closing of the ion-conducting pathway), for example, by increasing the sensitivity to Ca2+-induced activation of RyR2. A second group of molecules acts by controlling the movement of ions through the pore of the RyR2 channel, for example, by entering the pore and physically obstructing ion passage. A third group of compounds may alter RyR2 function by enhancing the interaction between subunits within the RyR2 macromolecular channel complex, or even between different RyR2 channel units (ie enhancing coupled gating). Although none of these compounds are currently used for the treatment of patients with heart failure or arrhythmias, the emphasis of this review will be on the potential therapeutic applications or non-therapeutic side effects of RyR2 modulating agents. Based on recent advances in our understanding of Ca2+-handling defects in heart failure and cardiac arrhythmias, one could profile an ideal drug for the modulation of RyR2. Such compounds would not interfere with systolic SR Ca2+ release, as this would depress cardiac contractility. However, inhibition of a diastolic SR Ca2+ leak would be desirable, as it is likely to prevent arrhythmias and enhances SR Ca2+ loading, which could improve contractility.
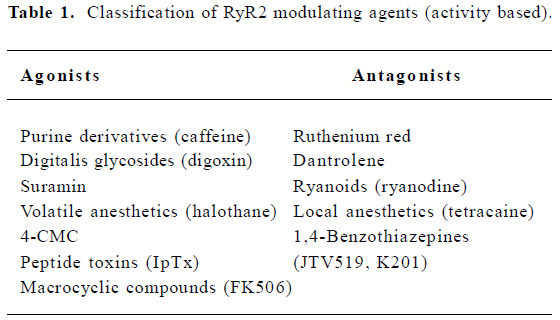
Full table
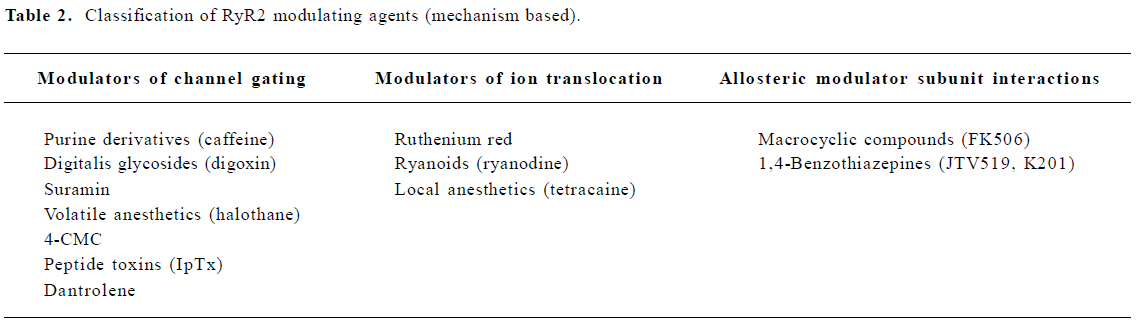
Full table
RyR2 agonists
Purine derivatives and related compounds This group includes substances that have a similar sterical structure based on a purine, carboline, carbazole, or imidazopyridine ring, and are likely to act on the same molecular site. Methylxanthine compounds, like caffeine and theophylline, isolated from the leaves and beans of certain plants, activate RyR2 by potentiating its sensitivity to the natural ligand Ca2+. RyR2 is activated by millimolar concentrations of caffeine, which causes a pronounced increase in the sensitivity of RyR2 to Ca2+ such that the channels open at basal (diastolic) Ca2+ levels[21]. At low caffeine concentrations, caffeine increases the open probability of the RyR2 channel by increasing the frequency of channel openings alone, whereas at higher concentrations, it results from an increase in both the open channel lifetime and the frequency of RyR2 openings[22]. Theophylline and other methylxanthines share the mode of action of caffeine[23]. Although these effects are readily observed in the experimental setting, it is unlikely that RyR2 modulation will be important in the therapeutic response to methylxanthines because their plasma concentration (eg about 55 micromolar for theophylline) is lower than the effective concentration range[20]. Further compounds have been proposed to act in a similar manner to caffeine. The imidazopyridine derivative, sulmazole, increases the duration and frequency of RyR2 openings. Whereas the EC50 for RyR2 activation by caffeine is between 0.2–1 millimolar, sulmazole displays much greater potency (400 µmol)[24,25].
Digitalis glycosides Digoxin is one of the cardiac glycosides, a closely-related group of drugs that have in common specific effects on the myocardium. These drugs are found in a number of plants; digoxin is extracted from the leaves of Digitalis lanata. At a therapeutic concentration (~1 nm), digoxin increases the open probability of RyR2 by decreasing the lifetime of the closed states of the channel[26]. Digoxin appears to sensitize RyR2, as channel gating itself is not modified at picomolar Ca2+ concentrations. The activation of RyR2, which is clearly distinct from the canonical Na+/K+-ATPase inhibiting action, might contribute to the inotropic effects of digoxin and digitoxin. Such actions are similar to those of caffeine and sulmazole, but digitalis glycosides do not affect the RyR1 isoform[27]. Owing to its strong effects on Na+/K+-ATPase, it is unlikely that digoxin will be used clinically to modulate RyR2 in the heart.
Suramin Suramin is a polysulphonated naphtylurea, originally developed for the treatment of trypanosomiasis and is also used as an anticancer agent. In single channel experiments, suramin (in micromolar concentrations) increases the open probability of sheep cardiac RyR2 channels by stabilizing the open conformational state[28]. Recently, it has been suggested that the complex changes in RyR2 activity may result from an interaction with CaM-binding sites[29]. Thus, the suramin-induced potentiation of Ca2+ release through RyR2 may involve a relief of CaM-induced inhibition. It is unclear at present whether suramin has any beneficial effects in animal models of heart failure.
Volatile anesthetics Several halogenated compounds affect SR Ca2+ release. The most extensively studied are volatile anesthetics such as halothane, and its isomer isoflurane. Halothane has been shown to increase SR Ca2+ release at gas concentrations ranging from about 0.002% to 3.8% (v/v) in a Ca2+- and pH-dependent manner[30,31]. At a physiological pH of 7.4, halothane increases RyR2 activity at all Ca2+ concentrations without affecting channel conductance[31,32]. Similar effects have been observed with isoflurane and enflurane (2.5%–4%). A reduction of the pH from 7.4 to 7.1 causes maximum channel activation to occur at much lower cytosolic Ca2+ concentrations[31]. Since the interaction of volatile anesthetics with RyR2 occurs at doses lower than the minimum effective alveolar concentration (ie ~0.7% for halothane and ~1.1% for isoflurane), their effects on RyR2 may produce negative inotropic effects and transient vasoconstriction[33]. Negative inotropy may result from enhanced diastolic Ca2+ release via RyR2, which reduces the levels of Ca2+ in the SR available for the subsequent systolic Ca2+ release. This in turn reduces the amplitude of the Ca2+ transient and suppresses cardiomyocyte contractility.
4-Chloro-m-cresol The phenol derivative 4-chloro-m-cresol (4-CMC) has been shown to increase the open probability of RyR1 incorporated in planar lipid bilayers by increasing both open lifetimes and frequencies[34]. In contrast to caffeine, 4-CMC can modulate channel gating from both the luminal and cytosolic sides of the channel. Whereas there are myriad data concerning the actions of 4-CMC on RyR1, relatively little is known about its action on the cardiac RyR2 isoform. In cell lines expressing recombinant RyR2, 4-CMC has been shown to enhance intracellular Ca2+ release[35]. Although 4-CMC may modulate RyR2, the significance of these pharmacological effects remains to be further explored.
Peptide toxins Several peptide toxins isolated from scorpion venoms have been shown to alter RyR2 activity, which has raised the prospect that animal venom may represent a unique source of selective modulators of intracellular Ca2+ release channels[36]. Two peptides isolated from the scorpion Pandinus imperator, imperatoxin A (IpTxa) and imperatoxin I (IpTxi), are highly selective for RyR and show no obvious activity with regard to other ion channels or transporters[37]. IpTxa is a small peptide comprising of 33 amino acids with a molecular weight of approximately 4 kDa. It specifically increases open probability of the RyR1 and RyR3 isoforms by sensitizing these channels to cytosolic Ca2+, but has little effect on RyR2. However, single channel experiments have revealed that IpTxa induces the occurrence of a subconductance state equivalent to ~30% of the full conductance in all RyR isoforms, even though IpTxa has no effect on RyR2 in ryanodine binding assays[38]. IpTxi is a larger heterodimeric protein (~15 kDa) that consists of a large subunit comprising of 104 amino acids covalently linked via a disulfide bond to a smaller subunit of 27 amino acids[39]. Single channel studies have demonstrated that IpTxi inhibits both RyR1 and RyR2 with nanomolar affinity, although these effects may be mediated via a lipid product of its inherent phospholipase-2 (PLA-2) activity[39]. In spite of these elegant electrophysiological data, in vivo pharmacological experiments are needed to determine whether IpTx can improve cardiac contractility in animals with heart failure.
Macrocyclic compounds The macrolide immunosuppressant FK-506, also known as tacrolimus, can induce the dissociation of FKBP12.6 from RyR2, thereby altering RyR2 gating. Rapamycin is another macrolide immunosuppressant that can dissociate FKBP12.6 from the RyR2 channel complex. In cardiac muscle, 0.1–10 µmol/L rapamycin increases single channel open probability and decreases channel conductance[40]. It has been speculated that the former effect is the consequence of drug binding to FKBP12.6, whereas the changes in channel conductance are the consequence of FKBP12.6 dissociation from RyR2[6,40]. Interference with the RyR2 subunit composition might be involved in some effects of FK506, particularly in the development of myocardial hypertrophy and heart failure, which has been observed in some pediatric transplant patients[41].
RyR2 antagonists
Ruthenium red Ruthenium red, a water-soluble dye with a structure that includes 14 amino groups, has been shown to inhibit SR Ca2+ release in cardiac muscle. In planar lipid bilayer experiments, micromolar concentrations of ruthenium red dramatically decreases RyR2 open probability, associated with long-term channel closure[42]. At submicromolar concentrations, the major effect of ruthenium red is a decrease in the lifetime of the open channel state, whereas at higher concentrations (>1 µmol/L), the lifetime of the closed channel is increased. Ruthenium red reduces the RyR2 single channel current from both the cytosolic and luminal sides of the channel. However, the dwell times of the block are longer when ruthenium red is added to the luminal side. In addition, luminal ruthenium red decreases the single channel current without affecting channel open probability[43]. Binding studies performed using recombinant RyR1 channels have localized ruthenium red binding sites at residues 1861–2094 and 3657–3776[44]. On the basis of single channel studies, it has been proposed that the drug-binding site is located within the transmembrane domain, probably close to the channel pore region, and ruthenium red cannot permeate through the open channel[43]. Because ruthenium red is neurotoxic, it is not an ideal candidate for drug development.
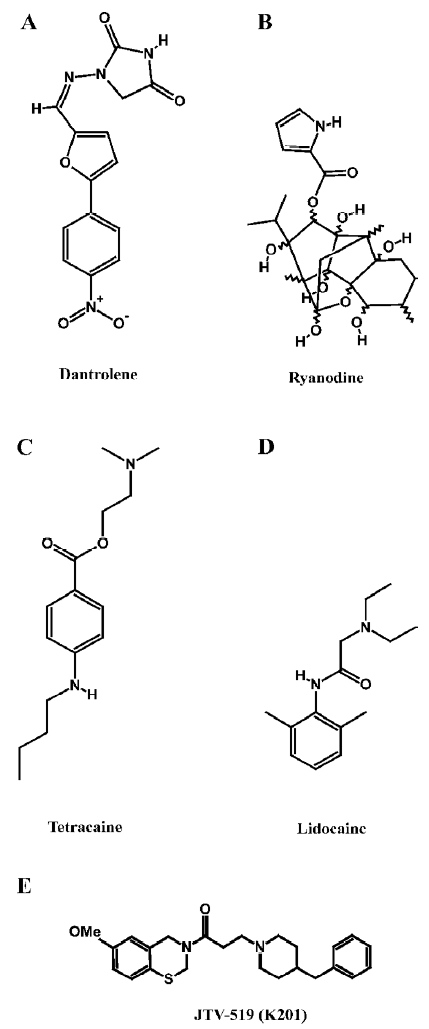
Dantrolene Dantrolene is a hydantoin derivative commonly used for the treatment of the genetic disorder, malignant hyperthermia, which is caused by inherited mutations in RyR1 (Figure 1A). Importantly, dantrolene represents the only drug targeting RyR that is currently approved for clinical use. In skeletal muscle, 10–100 micromolar dantrolene inhibits abnormal Ca2+ release from the SR[45]. The inhibition of SR Ca2+ release through RyR2 was also observed in cardiac muscle, but the sensitivity to dantrolene was lower than in the skeletal muscle. Recently, it has been demonstrated that dantrolene can stabilize domain–domain interactions within the RyR complex[46]. Taken together, these data suggest that dantrolene might exert therapeutic effects in heart failure by preventing an abnormal SR Ca2+ leak, although this has not been investigated yet in experimental models[19].
Ryanoids Ryanodine is a naturally-occurring plant alkaloid isolated from plants of the genus Ryania (Figure 1B). Because it binds with high affinity and specificity to its receptor in the SR, RyR have been named after this compound. Ryanodine is unusual in that it is a modulator of both gating and ion translocating properties of RyR2. The pharmacology of ryanodine has been described extensively in other literature reviews[20,47,48], therefore we will focus on its action on RyR2 and its potential application as a drug for the treatment of cardiovascular disorders.
RyR2 possess both a high- and low-affinity binding site for ryanodine, which contributes to the concentration-dependent effects of ryanodine on the activity of RyR. At nanomolar concentrations, ryanodine increases the open probability of RyR2 without affecting the rates of ion movement. At submicromolar concentrations, ryanodine increases RyR2 open probability to almost 100% and it induces a long-lasting subconductance state representing ~50% of the normal conductance level[47]. Finally, at high micromolar concentrations, ryanodine causes the channel to fully close, which accounts for an inhibitory effect on SR Ca2+ release[49].
Derivatives of ryanodine, collectively called ryanoids, display actions that do not conform to the canonical ryanodine characteristics. For example, some ryanoids can induce subconductance amplitudes far different from the half-open state (ranging from 6%–75% of the maximum amplitude)[47]. The duration of the subconductance states also vary considerably among ryanoids. The unique features of certain ryanoids could be considered when developing ryanodine derivatives for heart failure therapy for which RyR2 specificity, low-level subconductance, and reversibility may be desirable characteristics.
Local anesthetics Several charged local anesthetics are known to inhibit RyR2 channels. These include both tertiary amines (eg procaine, tetracaine, and lidocaine), as well as quaternary amines (eg QX572 and QX314)[50]. Although procaine and tetracaine (Figure 1C) are both effective at low millimolar concentrations, procaine appears to be more selective for RyR2 compared to RyR1. Single channel studies have revealed that both drugs decrease the RyR2 open probability by stabilizing a closed conformational state[51]. Single channel studies of RyR2 have revealed 2 different modes of action of local anesthetics. Tetracaine and procaine decreased the channel open probability by stabilizing a closed state of the channel without affecting its unitary conductance[51]. In contrast, lidocaine (Figure 1D) and quaternary amines appear to induce voltage-dependent channel blockade, characterized by reduced conductance without changes in the open probability. This voltage-dependent inhibition is also observed in the presence of millimolar concentrations of procaine or tetracaine, in the presence of 2 µmol/L ryanodine[51]. Interestingly, tetracaine has been shown to prevent arrhythmogenic spontaneous SR Ca2+ release events, presumably by reducing aberrant diastolic RyR2 openings[52]. Moreover, tetracaine also potentiates systolic Ca2+ release due to enhanced diastolic SR Ca2+ filling (due to decreased Ca2+ leak from the SR)[53]. Thus, compounds such as tetracaine may have a therapeutic benefit in the prevention of cardiac arrhythmias and contractile dysfunction in heart failure.
1,4-Benzothiazepines The pharmacological agents discussed earlier modulate RyR2 by directly altering channel gating or ion translocation. Recently, an additional mechanism for regulating RyR2 channels has been described[54,55]. It was shown that the 1,4-benzothiazepine derivative JTV519 (also known as K201, Figure 1E) stabilizes the interaction of RyR2 with the endogenous inhibitory subunit FKBP12.6[54-56]. The FK506-binding protein FKBP12.6 has previously been shown to stabilize a closed conformational state of the RyR2 channel, thereby decreasing the open probability[57]. In addition, JTV519 may enhance coupled gating between RyR2 channel complexes by increasing the binding of FKBP12.6[6]. Based on observations that FKBP12.6 binding to RyR2 is decreased in patients and animals with heart failure, the therapeutic role of JTV519 was assessed in disease models.
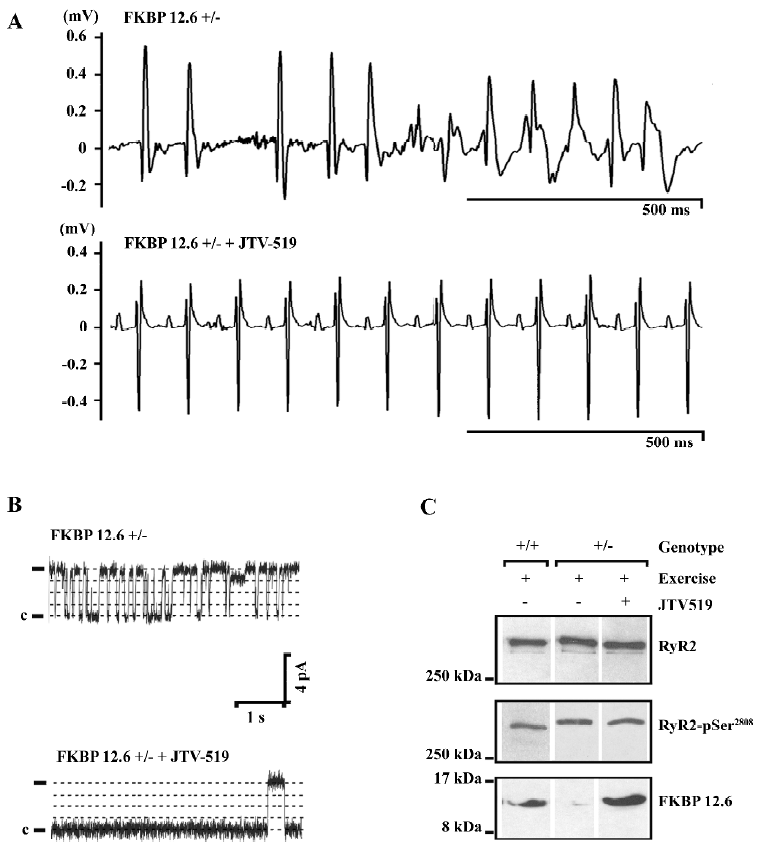
In animal models of heart failure and myocardial infarction, JTV519 has been demonstrated to improve contractile function and prevent the development of adverse left ventricular remodeling[54,56]. Because these therapeutic effects were not observed in FKBP12.6-deficient mice, it was concluded that these effects are dependent on the enhanced interaction of FKBP12.6 with RyR2[56]. Furthermore, it has been proposed that JTV519 may prevent cardiac arrhythmias that arise from delayed afterdepolarizations, initiated by a SR Ca2+ leak through FKBP12.6-depleted RyR[58,59]. JTV519 prevented catecholaminergic ventricular tachycardias in FKBP12.6 haploinsufficient mice, but not in FKBP12.6-deficient mice, again indicating that the enhanced binding of FKBP12.6 to RyR2 constitutes the therapeutic mechanism of this 1,4-benzothiazepine derivative (Figure 2)[19,60]. Although these animal studies are very promising, it remains to be seen whether or not JTV519 becomes a useful clinical drug in the treatment of cardiac arrhythmias and heart failure.
Conclusion
In this article, we provided a general overview of therapeutic approaches and pharmacological agents that are known to modulate RyR in the heart. Some of these compounds modulate channel gating, whereas others regulate ion translocation. The recent emergence of RyR2 as a critical defect in the pathogenesis of heart failure and triggered cardiac arrhythmias has spurred interest in developing novel therapeutic agents based on these channels. Although it is imperative that effective therapeutic agents do not interfere with systolic SR Ca2+ release (as this would depress cardiac contractility), the inhibition of a diastolic SR Ca2+ leak would be desirable as it likely prevents arrhythmias and enhances SR Ca2+ loading (thus improving contractility). Most of the classic RyR2 modulating drugs, however, display unacceptable side effects or lack long-term efficacy. The recently described 1,4-benzothiazepine JTV519, however, acts via a different mechanism, namely by allosteric modification of protein-protein interactions within the channel complex. Interestingly, this drug selectively targets diastolic SR Ca2+ leaks without affecting systolic Ca2+ release. Finally, these studies suggest that drugs that modify other subunits within the RyR2 macromolecular complex may also provide therapeutic benefits in patients with heart failure or arrhythmias.
References
- Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann N Y Acad Sci 1998;853:20-30.
- Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci 2005;1047:86-98.
- Du GG, MacLennan DH. Topology and transmembrane organization of ryanodine receptors. In: Wehrens XH, Marks AR, editors. Ryanodine receptors: structure, function and dysfunction in clinical disease; v 254. New York: Springer; 2005. p 9–23.
- Wehrens XHT, Lehnart SE, Marks AR. Intracellular calcium release channels and cardiac disease. Annu Rev Physiol 2005;67:69-98.
- Williams AJ, West DJ, Sitsapesan R. Light at the end of the Ca2+-release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q Rev Biophys 2001;34:61-104.
- Lehnart SE, Huang F, Marx SO, Marks AR. Immunophilins and coupled gating of ryanodine receptors. Curr Top Med Chem 2003;3:1383-91.
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 1993;262:740-4.
- Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med 2006;3:43-52.
- Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189-94.
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196-200.
- Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001;103:485-90.
- Wehrens XH. The molecular basis of catecholaminergic polymorphic ventricular tachycardia: what are the different hypotheses regarding mechanisms? Heart Rhythm 2007. in press.
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000;101:365-76.
- Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation 2000;102:2131-6.
- Phrommintikul A, Chattipakorn N. Roles of cardiac ryanodine receptor in heart failure and sudden cardiac death. Int J Cardiol 2006;112:142-52.
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phospho-rylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA 2006;103:511-8.
- Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res 2003;93:487-90.
- Bers DM. Cardiac ryanodine receptor phosphorylation: target sites and functional consequences. Biochem J 2006;396:1-3.
- Wehrens XH, Marks AR. Novel therapeutic approaches for heart failure by normalising calcium cycling. Nat Rev Drug Discov 2004;3:565-73.
- Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 1997;49:1-51.
- Meissner G. Adenine nucleotide stimulation of Ca2+-induced Ca2+ release in sarcoplasmic reticulum. J Biol Chem 1984;259:2365-74.
- Sitsapesan R, Williams AJ. Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J Physiol 1990;423:425-39.
- Seifert J, Casida JE. Ca2+-dependent ryanodine binding site: soluble preparation from rabbit cardiac sarcoplasmic reticulum. Biochim Biophys Acta 1986;861:399-405.
- McGarry SJ, Williams AJ. Activation of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by analogues of sulmazole. Br J Pharmacol 1994;111:1212-20.
- Tinker A, Sutko JL, Ruest L, Deslongchamps P, Welch W, Airey JA, et al. Electrophysiological effects of ryanodine derivatives on the sheep cardiac sarcoplasmic reticulum calcium-release channel. Biophys J 1996;70:2110-19.
- McGarry SJ, Williams AJ. Digoxin activates sarcoplasmic reticulum Ca(2+)-release channels: a possible role in cardiac inotropy. Br J Pharmacol 1993;108:1043-50.
- Sagawa T, Sagawa K, Kelly JE, Tsushima RG, Wasserstrom JA. Activation of cardiac ryanodine receptors by cardiac glycosides. Am J Physiol Heart Circ Physiol 2002;282:H1118-26.
- Sitsapesan R, Williams AJ. Modification of the conductance and gating properties of ryanodine receptors by suramin. J Membr Biol 1996;153:93-103.
- Hill AP, Kingston O, Sitsapesan R. Functional regulation of the cardiac ryanodine receptor by suramin and calmodulin involves multiple binding sites. Mol Pharmacol 2004;65:1258-68.
- Carrier L, Villaz M, Dupont Y. Abnormal rapid Ca2+ release from sarcoplasmic reticulum of malignant hyperthermia susceptible pigs. Biochim Biophys Acta 1991;1064:175-83.
- Beltran M, Bull R, Donoso P, Hidalgo C. Ca2+- and pH-dependent halothane stimulation of Ca2+ release in sarcoplasmic reticulum from frog muscle. Am J Physiol 1996;271:C540-6.
- Connelly TJ, Coronado R. Activation of the Ca2+ release channel of cardiac sarcoplasmic reticulum by volatile anesthetics. Anesthesiology 1994;81:459-69.
- Hanouz JL, Massetti M, Guesne G, Chanel S, Babatasi G, Rouet R, et al. In vitro effects of desflurane, sevoflurane, isoflurane, and halothane in isolated human right atria. Anesthesiology 2000;92:116-24.
- Herrmann-Frank A, Richter M, Sarkozi S, Mohr U, Lehmann-Horn F. 4-Chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochim Biophys Acta 1996;1289:31-40.
- George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res 2003;93:531-40.
- Gurrola GB, Zhu X, Valdivia HH. Scorpion peptides as high-affinity probes of ryanodine receptor function. In: Wehrens XH, Marks AR, editors. Ryanodine receptors: structure, function, and dysfunction in clinical disease. New York: Springer; 2005. p 191–200.
- Valdivia HH, Kirby MS, Lederer WJ, Coronado R. Scorpion toxins targeted against the sarcoplasmic reticulum Ca2+-release channel of skeletal and cardiac muscle. Proc Natl Acad Sci USA 1992;89:12185-9.
- Tripathy A, Resch W, Xu L, Valdivia HH, Meissner G. Impera-toxin A induces subconductance states in Ca2+ release channels (ryanodine receptors) of cardiac and skeletal muscle. J Gen Physiol 1998;111:679-90.
- Zamudio FZ, Conde R, Arevalo C, Becerril B, Martin BM, Valdivia HH, et al. The mechanism of inhibition of ryanodine receptor channels by imperatoxin I, a heterodimeric protein from the scorpion Pandinus imperator. J Biol Chem 1997;272:11886-94.
- Kaftan E, Marks AR, Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca2+ release channels from cardiac muscle. Circ Res 1996;78:990-7.
- Atkison P, Joubert G, Barron A, Grant D, Paradis K, Seidman E, et al. Hypertrophic cardiomyopathy associated with tacrolimus in paediatric transplant patients. Lancet 1995;345:894-6.
- Smith JS, Coronado R, Meissner G. Sarcoplasmic reticulum contains adenine nucleotide-activated calcium channels. Nature 1985;316:446-9.
- Ma J. Block by ruthenium red of the ryanodine-activated calcium release channel of skeletal muscle. J Gen Physiol 1993;102:1031-56.
- Chen SR, MacLennan DH. Identification of calmodulin-, Ca(2+)-, and ruthenium red-binding domains in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J Biol Chem 1994;269:22698-704.
- Parness J. The dantrolene binding site on RyR1. In: Wehrens XH, Marks AR, editors. Ryanodine receptors: structure, function and dysfunction in clinical disease. New York: Springer; 2005. p 243–51.
- Kobayashi S, Bannister ML, Gangopadhyay JP, Hamada T, Parness J, Ikemoto N. Dantrolene stabilizes domain interactions within the ryanodine receptor. J Biol Chem 2005;280:6580-7.
- Besch HR, Shao CH, Bidasee KR. Ryanoids, receptor affinity and RyR channel subconductance. In: Wehrens XH, Marks AR, editors. Ryanodine receptors: structure, function and dysfunction in clinical disease. New York: Springer; 2005. p 179–89.
- Sutko JL, Airey JA, Welch W, Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol Rev 1997;49:53-98.
- Nagasaki K, Fleischer S. Ryanodine sensitivity of the calcium release channel of sarcoplasmic reticulum. Cell Calcium 1988;9:1-7.
- Shoshan-Barmatz V, Zchut S. The interaction of local anesthetics with the ryanodine receptor of the sarcoplasmic reticulum. J Membr Biol 1993;133:171-81.
- Xu L, Jones R, Meissner G. Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. J Gen Physiol 1993;101:207-33.
- Overend CL, Eisner DA, O’Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. J Physiol 1997;502:471-9.
- Venetucci LA, Trafford AW, Diaz ME, O’Neill SC, Eisner DA. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res 2006;98:1299-305.
- Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 2003;107:477-84.
- Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 2004;304:292-6.
- Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA 2005;102:9607-12.
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 1994;77:513-23.
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003;113:829-40.
- Lehnart SE, Wehrens XHT, Laitinen PJ, Reiken SR, Deng SX, Chen Z, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 2004.113-9.
- Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA 2006;103:7906-10.