
Derivatives of aryl-4-guanidinomethylbenzoate and N-aryl-4-guanidinomethylbenzamide as new antibacterial agents: synthesis and bioactivity1
Introduction
The emergence of bacterial resistance to different classes of antibacterial agents, such as β-lactams, quinolones, and macrolides, is an alarming problem that seriously affects human health[1]. To combat this situation, numerous efforts have been made in the development of new approaches to treat bacterial infections, particularly for therapeutics with novel mechanisms of action and little or no cross-resistance[2–4]. As a result, new antibacterial agents against hospital-acquired Gram-positive bacterial pathogens[5], especially against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE), have become the center of attention in this highlighted research field[6,7].
The guanidino group has long been recognized as a ubiquitous moiety present in numerous therapeutic agents, including cardiovascular, antihistamine, anti-inflammatory, antidiabetic, antibacterial, antihypertensive, antiviral, and antineoplastic drugs[8]. Many natural products also contain guanidino moieties[9–12], and their strong cationic nature is often associated with biological activities. Of particular interest is the presence of guanidine in several peptidic antibiotics, such as capreomycin[13], viomycin[14], tuberac-tinomycin[15], and neomycin[16].
A class of guanidine-containing antibacterial agents that targets a proteinase of microbial origin was identified as competitive trypsin inhibitors in vitro and consists of various aromatic esters of trans-4-guanidinomethylcyclohexanecarboxylic acid (GMCHA)[17–20]. For instance, trans-4-guanidinomethyl cyclohexane carboxylic acid (4-[4-{4-methylbenzyloxy carbonyl}phenyl] phenylester) (TG44), a derivative of GMCHA[21], and 4-guanidinomethyl benzoic acid (4-[4-{4-methylbenzyloxy carbonyl}phenyl]phenylester) (NE-2001; Figure 1) are both selective synthetic antibiotic agents directed against Helicobacter pylori (H pylori )[22]. TG44, which is undergoing clinical development, possesses rapid bactericidal activity and is useful for eradicating not only the antibiotic-susceptible, but also the antibiotic-resistant strains of H pylori by monotherapy[21]. As a drug candidate, NE-2001 displays superior anti-H pylori efficacy on clinical isolates either resistant to metronidazole or resistant to both metronidazole and clarithromycin[22].
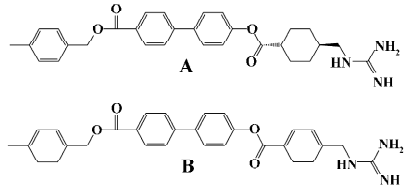
The activity of NE-2001 as a selective antibacterial agent against H pylori prompted us to conduct structural modifications of this core molecule. In this report, we describe the design, synthesis, and biological evaluation of novel guanidine derivatives of aryl-4-guanidinomethylbenzoate (series A) and N-aryl-4-guanidinomethylbenzamide (series B). Twelve of these compounds demonstrated potent antibacterial activities against Gram-positive microorganisms, such as Staphylococcus aureus and Staphylococcus epidermidis, but did not display or showed little or no activity against Gram-negative Escherichia coli, Pseudomonas aeruginosa, Salmonella typhi, and so on. The results suggest that these compounds may have the potential to be developed into narrow spectrum antibacterial agents to help control the wide spreading of drug resistance.
Materials and methods
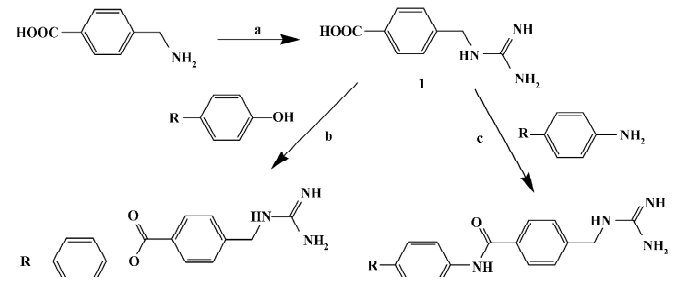
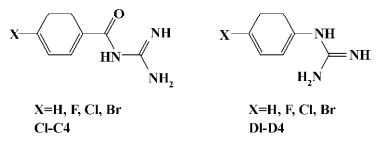
Chemistry General synthetic routes towards aryl-4-guanidinomethylbenzoate (A1–24) and N-aryl-4-guanidinomethylbenzamide derivatives (B1–20) are illustrated in Figure 2[23]. The target compound A6 was synthesized by condensation of 4-N-Boc-aminophenol and 4-guanidinomethylbenzoic acid (intermediate 1), followed by cleavage of the Boc group under acidic conditions[24]. The target compound B5 was synthesized by condensation of 4-benzyloxyaniline and intermediate 1, followed by cleavage of the benzyl group catalyzed with Pd-C[25]. Compounds A8–10 and B10–12 were prepared by condensation of the intermediate 1 with intermediates 4-halo-4'-hydroxybenzophenone or 4-amino-4'-halobenzophenone, respectively[26,27]. Compounds A12–14 and B14–16 were prepared by condensation of the intermediate 1 with intermediate 4-halo-4'-hydroxydiphenyl or 4-amino-4'-halodiphenyl, respectively[28–30]. Substituted benzoyl guanidines (C1–4) and substituted phenyl guanidines (D1–D4) were synthesized according to the literature[31,32]. 4-Guanidinobenzoic acid and 4-guanidinophenylacetic acid were synthesized by the reaction of aminoiminomethanesulfonic acid with 4-aminobenzoic acid and 4-aminophenylacetic acid, respectively[33], followed by condensation with 4-phenylphenol using dicyclohexylcarbodiimide (DCC) to give 4-phenylphenyl 4-guanidinobenzoate (E1) and 4-phenylphenyl 4-guanidinophenylacetate (E2)[23]. All of the compounds were converted to their corresponding hydrochloride salts for biological evaluation.
Antibacterial activity assay Antimicrobial activity was determined by an agar dilution method according to the Clinical and Laboratory Standard Institute (CLSI, formerly National Committee for Clinical Laboratory Standards, NCCLS) guidelines[34]. The test compounds were dissolved in DMSO and diluted at 1:2 serially in distilled water to produce various concentrations. The solutions were loaded onto plates and blundered with quantitative Mueller-Hinton agar medium. Inocula were prepared with overnight cultures and inoculated with a multipoint inoculator (MIT-P, Sakuma, Tokyo, Japan). Final inocula contained 1×104 colony-forming units (CFU)/spot. The plates were incubated at 35 °C for 18–24 h depending on the culture requirement. All the bacteria strains used in this study were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Meropenem dissolved in distilled water (Sigma-Aldrich, St Louis, MO, USA) was used as a positive control. Minimum inhibitory concentration (MIC) values were determined as the lowest concentration of the compound that inhibited visible growth.
Enzyme inhibition assay The oligopeptidase B (OpdB) expression vector pET28a-OpdB was provided by Dr Xue-yuan JIANG, Department of Biochemistry, Nanjing University (Nanjing, China). Expression, purification, and the enzymatic inhibition assay were carried out as described previously[35] with minor modifications. The reaction volume was reduced to 100 µL from 2 mL in which the amount of enzyme, substrate, and test compounds decreased, but their concentrations remained unchanged. Boc-Glu-Lys-Lys-MCA (Peptide Institute, Osaka, Japan) was used as the substrate for OpdB with a final concentration in the reaction mixture of 10 µmol/L. The liberated 4-methylcoumaryl-7-amide (MCA) was measured fluorometrically with the EnVision 2101 multilabel reader (PerkinElmer, Boston, MA, USA). The excitation and emission wavelengths were 355 and 460 nm, respectively.
Purified OpdA (powder) was also supplied by Dr JIANG and the assay procedure to measure OpdA activity was similar to that in the literature[35], except that the reaction volume was reduced to 100 µL. Enzymatic activities were determined fluorometrically with a Shimadzu RF-5301 spectrofluorometer (Shimadzu Corporation, Kyoto, Japan).
Data analysis Data were analyzed using GraphPad Prism software (San Diego, CA, USA).
Results
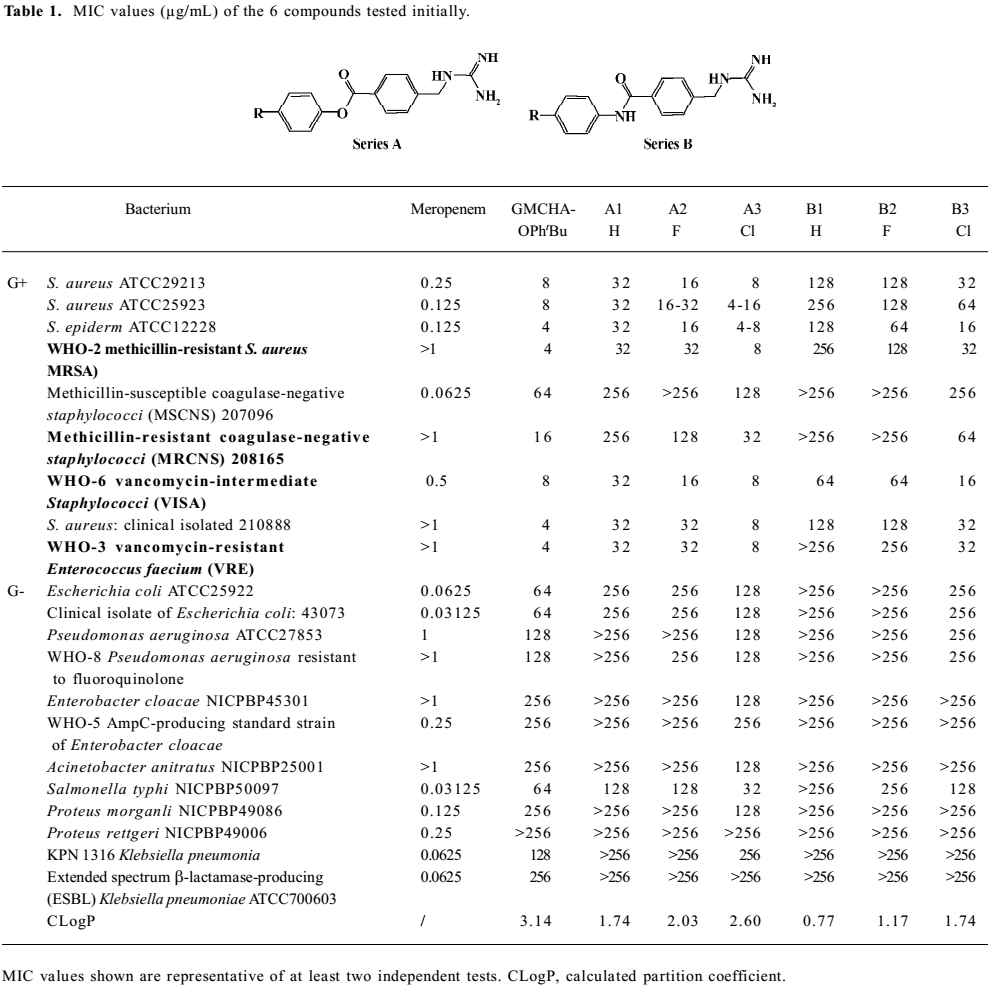
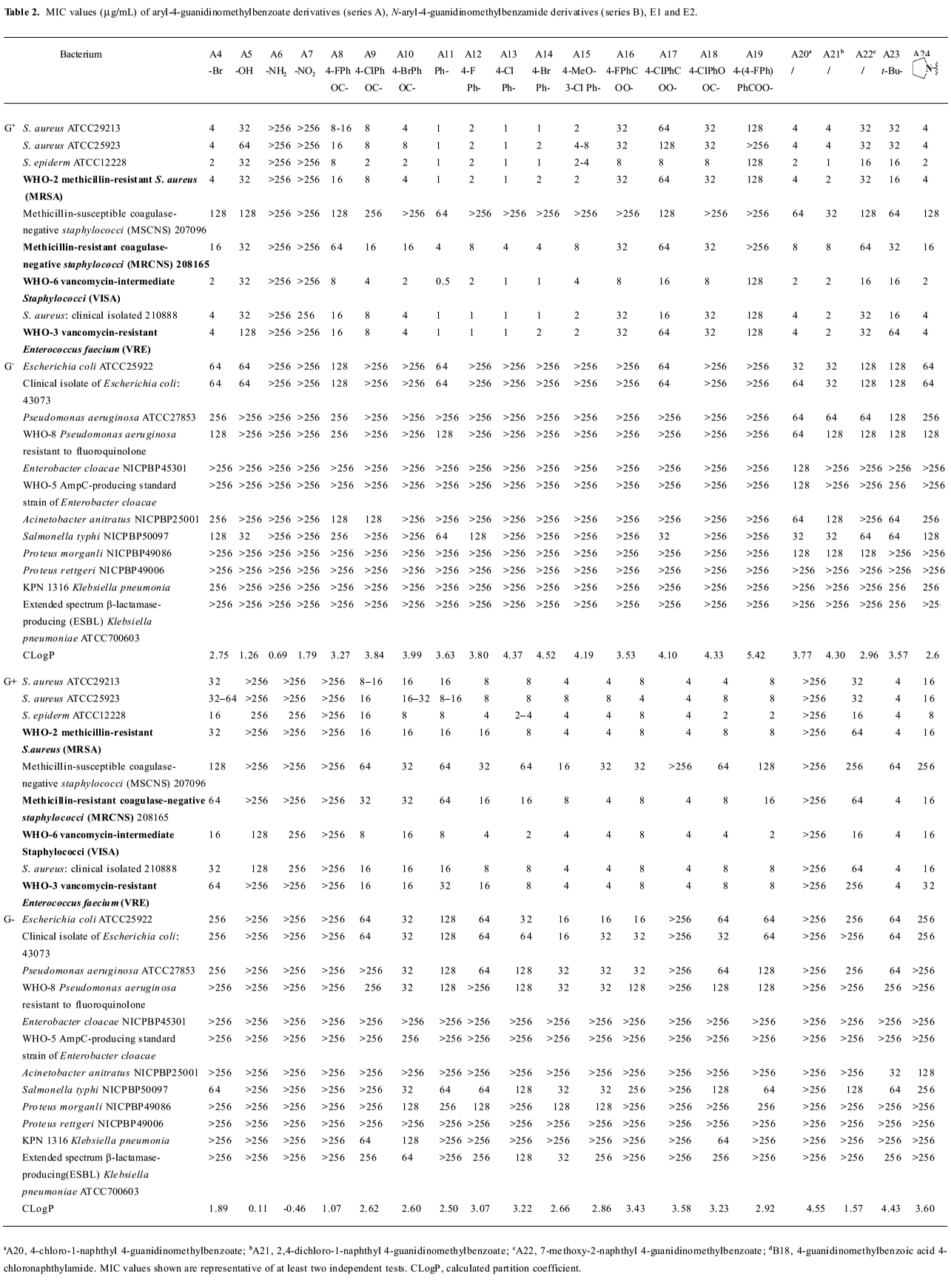
Growth inhibitory activities against Gram-positive and negative bacteria We first observed moderate antibacterial activities to certain Gram-positive bacteria, and very little effect on some Gram-negative bacteria in 6 of the series A and B compounds (Table 1). This phenomenon was abolished when substituted benzoyl guanidines (C1−4) and substituted phenyl guanidines (D1−4) were tested (MIC>256 µg/mL; Figure 3). Subsequent structural modifications were directed towards a variety of substitutions on the phenyl group, which led to the synthesis of 2 series of compounds, aryl-4-guanidinomethylbenzoate derivatives (series A) and N-aryl-4-guanidinomethylbenzamide derivatives (series B; Table 2).
Full table
Full table
It was noted that many of the compounds (series A in particular) were highly selective against most Gram-positive microorganisms tested (eg Staphylococcus aureus ATCC29213, Staphylococcus aureus ATCC25923, Staphylococcus epidermidis ATCC12228), while their inhibitory effects on a range of Gram-negative bacteria were minimal (eg Escherichia coli ATCC25922, Pseudomonas aeruginosa ATCC27853, Enterobacter cloacae NICPBP45301, Salmonella typhi NICPBP50097, Acinetobacter anitratus NICPBP25001, Proteus morganli NICPBP49086, Proteus rettgeri NICPBP49006, Shigella dysenteriae, and Shigella flexneri). This lack of efficacy may be attributed to their poor ability to penetrate the additional outer membrane barrier of Gram-negative bacteria[36].
In addition to the Gram-positive and -negative bacteria tested, these compounds also exhibited bactericidal properties against Bacillus cereus (MIC=4–32 µg/mL), Bacillus subtilis (MIC=2–16 µg/mL), Mycobacterium phlei (MIC=4–32 µg/mL), and Candida albicans (MIC =8–32 µg/mL).
Interestingly, the 2 series of compounds were highly efficacious in suppressing the growth of drug-resistant strains of Gram-positive bacteria, including MRSA, VRE, vancomycin-intermediate Staphylococci (VISA), and methicillin-resistant coagulase-negative Staphylococci (MRCNS; Table 3). The effects on the 4 strains of drug-resistant bacteria were similar to that observed with drug-susceptible strains of bacteria such as Staphylococcus aureus ATCC 29213, Staphylococcus aureus ATCC 25923, and Staphylococcus epidermidis ATCC12228 (Table 2).

Full table
The above results (Tables 1,2) allowed us to draw some conclusions. The introduction of halogen atoms resulted in a reasonable inhibitory activities, whereas compounds lacking substitutes (A1 and B1) are less active. We also attempted to increase the aqueous solubility of the compounds by introducing amino and hydroxyl groups on the phenyl group, as shown with compounds A5–6 and B5–6, but we observed a drastic decrease in their antibacterial effects. However, substitution by the hydrophobic groups seemed to improve the in vitro MIC values. The benzyloxy group (B8) brought about moderate activity.
Further introduction of a benzoyl group to the phenyl group, giving benzophenone derivatives, elicited better in vitro MIC values for compounds A8–10 and B10–12. Substitution by the naphthyl group also improved activity (A20–21 and B18). The modification of the phenyl ring to a diphenyl ring (A11–15 and B13–17) demonstrated the most potent antibacterial activity against drug-resistant Gram-positive bacteria with MIC values of 0.5–8 µg/mL. This may be attributed to the hydrophobic nature of the diphenyl ring compared to the benzophenone derivatives. This also indicates that it is favorable to have the hydrophobic group at the phenyl site, and bulky hydrophobic groups increase the activity. Compounds A11 and B13, which were substituted by a phenyl group, displayed more than a 32-fold increase in bioactivity in comparison with A1 and B1. A12–14 and B14−16 with 4-halophenyl substitutions demonstrated a 4- to 16-fold increase in antibacterial effects as opposed to halogen-substituted phenyl-4-guanidinomethylbenzoate derivatives. Series A compounds possessed much better bactericidal efficacy and selectivity against Gram-positive strains than their counterparts in series B substituted with the same group. We also synthesized 2 guanidinophenyl derivatives (E1−2; Table 2), but their activities were reduced compared to A11, indicating that the methylene group was required in the series A core structure.
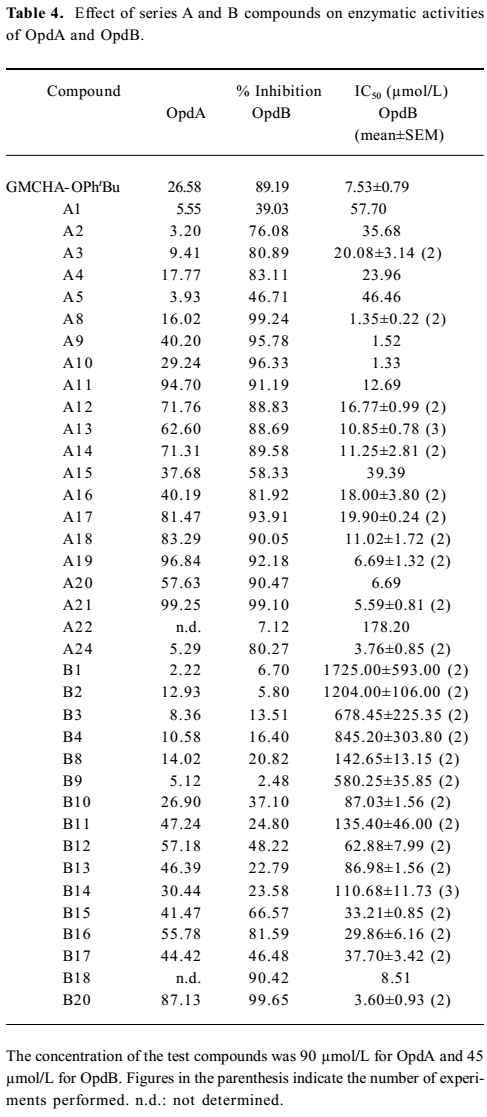
Inhibitory activity against OpdA and OpdB We next evaluated the inhibitory effects of designed compounds on 2 trypsin-like proteinases, oligopeptidase A (OpdA) and OpdB isolated from Escherichia coli[35]. Table 4 summarizes the inhibitory percentage and the 50% inhibition concentration (IC50) values against OpdA and OpdB of a majority of the synthesized compounds with MIC values (against Staphylococcus aureus ATCC25923) lower than 256 µg/mL. It is remarkable that many compounds showed high inhibitory effects (low IC50 values) against OpdB. We further studied the correlation between the MIC values and the inhibitory effects on OpdB of the series A and B compounds with basically similar substitutions. A general correlation could be established between the antibacterial activities (MIC50 values of the compounds against Staphylococcus aureus ATCC25923) and inhibitory effects on OpdB (IC50; Figures 4,5). However, we found that the benzophenone derivatives (A8–10) did not display this tendency (Figure 4) while possessing fairly low IC50 values (Table 4). The reason of this apparent discrepancy between bactericidal activity and the inhibition on OpdB remains to be investigated. However, there was a good correlation between the MIC and IC50 values for all the Series B compounds studied (Figure 5). No similar trend was found with OpdA.
Full table


Discussion
The growing incidence and frequency of bacterial resistance to current therapeutic agents remains a huge challenge for infectious disease specialists and pharmaceutical companies. In order to keep ahead of this growing issue, novel compounds working by new mechanisms of action are required. Topping the list of infections of most concern are MRSA and VRE. As an example, using the baseline data from 1997, it is shown that in 2002 Staphylococcus aureus was the most frequently diagnosed pathogen and exhibited significantly less susceptibility to a variety of antibacterial agents, clearly indicating the need for continued efforts to expand the arsenal of drugs available[37].
It was reported previously that 4-guanidinomethylbenzoic acid arylamides had potent inhibitory effects on gastric lesions[38]. However, their bactericidal effects were not examined. In this study, we describe a novel class of synthetic antimicrobial agents based on a similar structural core (aryl-4-guanidinomethylbenzoate and N-aryl-4-guanidinomethylbenzamide derivatives) with a narrow spectrum against Gram-positive bacteria, including several drug-resistant strains of clinical importance.
A preliminary structure-activity relationship (SAR) analysis was performed to identify the active groups on both series A and B compounds. Substitution by the hydrophobic groups on the phenyl ring increases the antibacterial activity, and bulky hydrophobic groups further contribute to this effect. Series A compounds possessed much better bactericidal efficacy and selectivity against Gram-positive strains than their counterparts in series B.
Linezolid, vancomycin, ciprofloxacin, levofloxacin, gemifloxacin, quinupristin-dalfopristin, and teicoplanin are common antimicrobial agents used in clinics. Linezolid resistance (MIC=16–32 µg/mL) was reported recently in clonally vancomycin-susceptible and -resistant Enterococcus faecium isolated from a patient after only 12 days of therapy[39], which indicates the urgency of finding alternative solutions. Table 3 compares the activities of 14 compounds (8 from series A and 6 from series B) described in this study with a number of antibiotic agents reported in the literature[40–44]. The antibacterial activities of A11–15 and A21 are superior to that of ciprofloxacin against MRSA and VRE. All series A and B compounds listed display comparable or better efficacy than ciprofloxacin and levo-floxacin against MRSA, VRE, and MRCNS, and they are sensitive and effective on VISA. With reference to the 4 drug-resistant strains, gemifloxacin shows relatively higher potency than most series A and B compounds. A11–15 and A21 (i) have comparable efficacies as eperezolid, but are slightly less active than quinupristin-dalfopristin; (ii) exhibit an equal potency as linezolid on MRSA, VRE, and VISA; (iii) show similar MIC values as vancomycin and teicoplanin against MRSA, and are more effective than vancomycin and teicoplanin on VRE; and (iv) demonstrate lower potency to MRCNS than most of the antibiotics compared. However, B13–18 are in general less efficacious than A11–15 and A21 (MIC=4–16 µg/mL).
The core structure of both series A and B compounds were designed based on NE-2001, whose putative target may involve a proteinase of microbial origin[22]. OpdA (EC 3.4.24.70) is the major soluble enzyme in Escherichia coli capable of hydrolyzing the free lipoprotein signal peptide in vitro[45]. As a member of the Zn metalloprotease subfamily[46], it is required for the normal development of phage P22[47] and can degrade the cleaved lpp signal peptide in vitro[48]. OpdB (EC 3.4.21.83) is a member of the prolyl oligopeptidase family of serine peptidases belonging to clan SC, family S9. It is a trypsin-like proteinase commonly found in ancient eukaryotic unicellular organisms, Gram-negative bacteria, and spirochetes[49]. Similar enzymes also exist in some plants and higher organisms[50,51] and have been implicated in the pathogenicity of certain bacteria since the enzyme is involved in host cell invasion by acting as an important virulence factor[49,52]. OpdB from Trypanosoma brucei has been identified as a target of several drugs used to treat African trypanosomiasis. The role of OpdB in the pathogenesis of several parasitic diseases and the possibility that OpdB represents a novel target for antimicrobial chemotherapy prompted an analysis of OpdB homologues from bacterial pathogens[53]. Although OpdB was not found in Gram-positive bacteria, the possibility exists that a surrogate molecule may exert similar functions. It is known that OpdB generates a calcium-signaling factor that interacts with a receptor on the mammalian cell surface, mobilizing Ca2+ from intracellular pools and promoting invasion by Trypanosoma cruzi. The targeted deletion of OpdB in Trypanosoma cruzi causes significant impairment of their ability to infect mammalian cells[54,55]. Therefore, this enzyme has the potential of becoming a therapeutic target. Indeed, the trypanocidal action of several drugs is highly correlated with their inhibitory effects on OpdB of a suramin analogue[56].
The results presented in Table 4 demonstrate that, similar to 4-tert-butylphenyl ester of GMCHA (GMCHA-OPhtBu; Figure 6)[35], a synthetic trypsin inhibitor, both series A and B compounds were capable of inhibiting enzymatic activities of OpdA and OpdB to various extents, but they were in general more selective towards OpdB. When comparing the antibacterial effects (MIC values) of both series A and B compounds with their ability to inhibit OpdB, a clear correlation could be established (Figures 4, 5). This trend was particularly evident with series A and B compounds substituted by small groups, such as halo- (A2–4 and B2–4), hydroxyl- (A5), t-butyl- (B9) and halophenyl- (A12–A15 and B14–17) groups. When substitutions were made with larger or complex groups, such as 4-haloben-zoyloxy (A16 and A17), 4-chlorophenyloxycarbonyl (A18), and 4-(4-fluorophenyl)benzoyloxy (A19), the correlation between MIC and OpdB inhibition was diminished. The analysis leads to our speculation that derivatives of aryl-4-guanidinomethylbenzoate and N-aryl-4-guanidino-methylbenzamide may target a putative OpdB-like molecule in Gram-positive germs to exert antibacterial actions, although an alternative possibility, that is, the compounds are acting as detergents and their activities are due to lysis of the cell membrane of the Gram-positive bacteria, cannot be ruled out at present.

In summary, we have designed, synthesized, and evaluated the antibacterial activities of aryl-4-guanidinomethyl-benzoate and N-aryl-4-guanidinomethylbenzamide derivatives. Of the compounds tested, A11–15, A20, and A21 displayed consistent and superior antibacterial effects against a spectrum of Gram-positive microorganisms, but showed little or no inhibitory activity against Gram-negative bacteria. Most of these compounds had comparable in vitro bactericidal activities against VRE and VISA as linezolid: their growth inhibitory effects on MRSA were similar to vancomycin, but were less potent than linezolid and vancomycin against MRCNS. The N-aryl-4'-guanidinomethylbenzamide derivatives B13–17 exhibited similar properties, but were less selective and efficacious. A preliminary SAR analysis was performed to identify the active groups on these compounds, and the mechanism of actions may involve the inhibition of a putative OpdB-like molecule in Gram-positive bacteria.
Acknowledgements
We are indebted to Ms Jie-ying ZHANG for technical assistance and to Dr Dale E MAIS for critical review of this manuscript.
References
- Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev 2005;105:395-424.
- Sandhya RG, McColm J, Clements JM, Taupin P, Barrowcliffe S, Hevizi S, et al. Pharmacokinetics in animals and humans of a first-in-class peptide deformylase inhibitor. Antimicrob Agents Chemother 2004;48:4835-42.
- Jarvest RL, Berge JM, Berry V, Boyd HF, Brown MJ, Elder JS, et al. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against Gram-positive pathogens. J Med Chem 2002;45:1959-62.
- Dyatkina NB, Roberts CD, Keicher JD, Dai Y, Nadherny JP, Zhang W, et al. Minor groove DNA binders as antimicrobial agents. Pyrrole tetraamides are potent antibacterials against vancomycin resistant Enteroccoci and methicillin resistant Staphylococcus aureus. J Med Chem 2002;45:805-17.
- Stefano D, Letizia B, Stefania S, Margherita S, Sofia S. Discovering novel antibacterial agents by high throughput screening. Front Drug Des Discov 2005;1:3-16.
- Ko KS, Kim YS, Song JH, Yeom JS, Lee H, Jung SI, et al. Genotypic diversity of methicillin-resistant Staphylococcus aureus isolates in Korean hospitals. Antimicrob Agents Chemother 2005;49:3583-5.
- Raad II, Hanna HA, Hachem RY, Dvorak T, Arbuckle RB, Chaiban G, et al. Clinical-use-associated decrease in susceptibility of vancomycin-resistant Enterococcus faecium to linezolid: a comparison with quinupristin–dalfopristin. Antimicrob Agents Chemother 2004;48:3583-5.
- Greenhill JV, Lue P. Amidines and guanidines in medicinal chemistry. Prog Med Chem 1993;30:203-326.
- Berlinck RG, Kossuga MH. Natural guanidine derivatives. Nat Prod Rep 2005;22:516-50.
- Berlinck RG. Natural guanidine derivatives. Nat Prod Rep 2002;19:617-49.
- Berlinck RG. Natural guanidine derivatives. Nat Prod Rep 1999;16:339-65.
- Berlinck RG. Natural guanidine derivatives. Nat Prod Rep 1996;13:377-409.
- Bycroft BW, Cameron D, Johnson AW. Synthesis of capreomycidine and epicapreomycidine, the epimers of alpha-(2-iminohexahydropyrimid-4-yl)glycine. J Chem Soc C 1971.3040-7.
- Bycroft BW, Croft LR, Johnson AW, Webb T. Viomycin. Part I. The structure of the guanidine-containing unit. J Chem Soc Perkin Trans 1 1972.820-7.
- Yoshioka H, Aoki T, Goko H, Nakatsu K, Noda T, Sakakibara H, et al. Chemical studies on tuberactinomycin. II. The structure of tuberactinomycin 0. Tetrahedron Lett 1971;12:2043-6.
- Schwab LE, Garner OB, Schuksz M, Crawford BE, Esko JD, Tor Y. Guanidinylated neomycin delivers large, bioactive cargo into cells through a heparan sulfate-dependent pathway. J Biol Chem 2007;282:13585-91.
- Kato M, Irisawa T, Ohtani M, Muramatu M. Purification and characterization of proteinase In: a trypsin-like proteinase, in Escherichia coli. Eur J Biochem 1992;210:1007-14.
- Kato M, Irisawa T, Morimoto Y, Muramatu M. A trypsin inhibitor trans-4-guanidinomethylcyclohexanecarboxylic acid 4-tert-butylphenyl ester suppresses the onset of DNA synthesis in Escherichia coli cells synchronized by phosphate starvation. Biol Pharm Bull 1993;16:552-7.
- Kato M, Irisawa T, Morimoto Y, Muramatu M. Effects of synthetic trypsin inhibitors on the growth of Escherichia coli. Biol Pharm Bull 1993;16:120-4.
- Irisawa T, Kato M, Moroishi J, Muramatu M. Effect of trans-4-guanidinomethylcyclohexanecarboxylic acid 4-tert-butylphenyl ester, a trypsin inhibitor, on the growth of various strains of Escherichia coli. Biol Pharm Bull 1993;16:621-6.
- Kamoda O, Anzai K, Mizoguchi J, Shiojiri M, Yanagi T, Nishino T, et al. In vitro activity of a novel antimicrobial agent, TG44, for treatment of Helicobacter pylori infection. Antimicrob Agents Chemother 2006;50:3062-9.
- Dai GF, Cheng N, Dong L, Muramatsu M, Xiao SD, Wang MW, et al. Bactericidal and morphological effects of NE-2001, a novel synthetic agent directed against Helicobacter pylori. Antimicrob Agents Chemother 2005;49:3468-73.
- Zhu DX, Muramatsu M, Xie JS, Cheng N, Wang MW. Methods and compositions for treating or preventing bacterial infection. US Patent 6 734 212. 2004; May 11.
- Yamashiro D, Blake J, Li CH. Use of N. alpha., Nim-bis(tert-butyloxycarbonyl)histidine and N.alpha.-2-(p-biphenylyl)isopropyloxycarbonyl-Nim-tert-butyloxycarbonylhistidine in the solid-phase synthesis of histidine-containing peptides. J Am Chem Soc 1972;94:2855-9.
- Johnstone RW, Wilby AH, Entwistle ID. Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. Chem Rev 1985;85:129-70.
- Pews RG, Tsuno Y, Taft RW. A fluorine nuclear magnetic resonance shielding study of substituent effects on charge distributions in benzophenone and its Lewis acid adducts. J Am Chem Soc 1967;89:2391-6.
- Kubo K, Ohyama SI, Shimizu T, Takami A, Murooka H, Nishitoba T, et al. Synthesis and structure-activity relationship for new series of 4-phenoxyquinoline derivatives as specific inhibitors of platelet-derived growth factor receptor tyrosine kinase. Bioorg Med Chem 2003;11:5117-33.
- Lemenovskii DA, Makarov MV, Dyadchenko VP, Bruce AE, Bruce MR, Larkin SA, et al. Syntheses and crystal structures of ferrocenyl derivatives of biphenyl. Rus Chem Bull 2003;52:607-15.
- Belcher R, Nutten AJ, Stephen WI. Substituted benzidines and related compounds as reagents in analytical chemistry. Part XII. Reagents for the precipitation of sulphate. J Chem Soc 1953.1334-8.
- Roe A, Fleishman HL. The synthesis of some fluorohydroxy-biphenyls. J Am Chem Soc 1947;69:509-10.
- Laeckmann D, Rogister F, Dejardin JV, Christelle PM, Géczy J, Delarge J, et al. Synthesis and biological evaluation of aroylguanidines related to amiloride as inhibitors of the human platelet Na+/H+ exchanger. Bioorg Med Chem 2002;10:1793-804.
- Scott FL, O’Donovan DG, Reilly J. Studies in the pyrazole series III. Substituted guanidines. J Am Chem Soc 1953;75:4053-4.
- Miller AE, Bischoff JJ. A facile conversion of amino acids to guanidine acids. Synthesis 1986;9:777-9.
- National Committee for Clinical Laboratory Standards. Performance standards for antimicrobial susceptibility testing and approved standard M7-A6. Informational supplement M100-S15 (2005). National Committee for Clinical Laboratory Standards: Wayne, PA, USA; 2003.
- Yan JB, Wang GQ, Du P, Zhu DX, Wang MW, Jiang XY. High-level expression and purification of Escherichia coli oligopeptidase B. Protein Expr Purif 2006;47:645-50.
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 2003;67:593-656.
- Fritsche TR, Stephen JS, Jones RN. Changing patterns of bacterial pathogens and their antimicrobial susceptibility profiles in patients hospitalized with pneumonia. Report from the SENTRY Antimicrobial Surveillance Program, 2002. Program and abstracts of the 43rd Annual ICAAC; Sep 14–17 2003, Illinois, Chicago, USA; Abstract C-2191.
- Ueda H, Ikeda T, Kakegawa H, Miyataka H, Matsumoto H, Satoh T. Syntheses and inhibitory effects on gastric lesions of 4-guanidinomethylbenzoic acid arylamides. Chem Pharm Bull 1993;41:1387-90.
- Seedat J, Zick G, Klare I, Konstabel C, Weiler N, Sahly H. Rapid emergence of resistance to linezolid during linezolid therapy of an Enterococcus faecium infection. Antimicrob Agents Chemother 2006;50:4217-9.
- Rybak MJ, Cappelletty DM, Moldovan T, Aeschlimann JR, Kaatz GW. Comparative in vitro activities and postantibiotic effects of the oxazolidinone compounds eperezolid (PNU-100592) and linezolid (PNU-100766) versus vancomycin against staphylococcus aureus, coagulase-negative staphylococci, Enterococcus faecalis, and Enterococcus faecium. Antimicrob Agents Chemother 1998;42:721-4.
- Howe RA, Wootton M, Noel AR, Bowker KE, Walsh TR, MacGowan AP. Activity of AZD2563, a novel oxazolidinone, against staphylococcus aureus strains with reduced susceptibility to vancomycin or linezolid. Antimicrob Agents Chemother 2003;47:3651-2.
- Tanaka M, Yamazaki E, Chiba M, Yoshihara K, Akasaka T, Takemura M. In vitro antibacterial activities of DQ-113, a potent quinolone, against clinical isolates. Antimicrob Agents Chemother 2002;46:904-8.
- Bogdanovich T, Esel D, Kelly LM, Bozdogan B, Credito K, Lin G, et al. Antistaphylococcal activity of DX-619, a new des-F(6)-quinolone, compared to those of other agents. Antimicrob Agents Chemother 2005;49:3325-33.
- Park HS, Kim HJ, Seol MJ, Choi DR, Choi EC, Kwak JH. In vitro and in vivo antibacterial activities of DW-224a, a new fluoronaph-thyridone. Antimicrob Agents Chemother 2006;50:2261-4.
- Novak P, Ray PH, Dev IK. Localization and purification of two enzymes from Escherichia coli capable of hydrolyzing a signal peptide. J Biol Chem 1986;261:420-7.
- Conlin CA, Vimr ER, Miller CG. Escherichia coli prlC encodes an endopeptidase and is homologous to the Salmonella typhimurium opdA gene. J Bacteriol 1992;174:5869-87.
- Novak P, Dev IK. Degradation of a signal peptide by protease IV and oligopeptidase A. J Bacteriol 1988;170:5067-75.
- Conlin CA, Miller CG. Dipeptidyl carboxypeptidase and oligopeptidase A from Escherichia coli and Salmonella typhimurium. Methods Enzymol 1995;248:567-79.
- Morty RE, Fulop V, Andrews NW. Substrate recognition properties of oligopeptidase B from Salmonella enterica serovar typhimurium. J Bacteriol 2002;184:3329-37.
- Tsuru D, Yoshimoto T. Oligopeptidase B: protease II from Escherichia coli. Methods Enzymol 1994;244:201-15.
- Tsuji A, Yuasa K, Matsuda Y. Identification of oligopeptidase B in higher plants. Purification and characterization of oligopeptidase B from quiescent wheat embryo, triticum aestivum. J Biochem 2004;136:673-81.
- Tsuji A, Yoshimoto T, Yuaask K, Matsuda Y. Protamine: a unique and potent inhibitor of oligopeptidase B. J Peptide Sci 2006;12:65-71.
- Tünde J, Zoltán S, Veronika R, László P. Role of the oxyanion binding site and subsites S1 and S2 in the catalysis of oligopeptidase B, a novel target for antimicrobial chemotherapy. Biochemistry 2002;41:4096-106.
- Morty RE, Authi’e E, Troeberg L, Lonsdale-Eccles JD, Coetzer THT. Purification and characterization of a trypsin-like serine oligopeptidase from Trypanosoma congolense. Mol Biochem Parasitol 1999;102:145-55.
- Morty RE, Lonsdale-Eccles JD, Mentele R, Auerswald EA, Coetzer THT. Trypanosoma-derived oligopeptidase B is released into the plasma of infected rodents, where its persists and retains full catalytic activity. Infect Immun 2001;69:2757-61.
- Morty RE, Troeberg L, Pike RN, Jones R, Nickel P, Lonsdale-Eccles JD, et al. A trypanosome oligopeptidase as a target for the trypanocidal agents pentamidine, diminazone and suramin. FEBS Lett 1998;433:251-6.