
Tetrandrine suppresses lipopolysaccharide-induced microglial activation by inhibiting NF-κB pathway1
Introduction
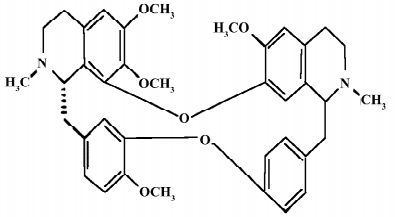
The Chinese herb Han-Fang-Ji (Stephania tetrandra S Moore) has been used in the therapy of rheumatic disorders for a long time in China[1]. The main bioactive component tetrandrine (TET) is a bisbenzylisoquinoline alkaloid with anti-inflammatory, anti-allergic, antioxidant, antifibrogenic, and antithrombogenic properties[2–5]. TET has been used to treat patients with silicosis, autoimmune disorders, and hypertension for decades. The antitumor properties of TET have also been demonstrated in various cancer models. The chemical structure of TET is illustrated in Figure 1.
Recently, in vitro research has shown that TET inhibits cellular proliferation through the suppression of cytokine production including interferon-γ, interleukin (IL)-4, IL-2, and IL-10. TET also inhibited IκBα kinases and protein kinase C activity[6,7], and prevented tumor necrosis factor-α (TNF-α) production in activated monocytes[8]. In addition, TET was found to have an inhibitory effect on calcium-dependent TNF-α production in glia-neuron mixed cultures[9].
Microglia comprise 12% of the cell population in the central nervous system (CNS)[10], and exist in multiple morphological states in healthy and injured brains. Microglia express surface markers similar to those of macrophages in peripheral tissues, thus are macrophage-like cells resident within the CNS. Moreover, microglia can mediate the initiation of inflammation in the CNS. Recent experimental evidence demonstrates that inflammation mediated by microglia contributes to neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease[11–13]. Activated microglia were shown to be capable of releasing various molecules, such as nitric oxide (NO), superoxide anion (O2–), IL-6, and TNF-α. Although NO was reported to play a role in the protection of neuron apoptosis, it was likely that NO at higher concentrations, mainly generated by inducible NO synthase (iNOS), exerted their detrimental effects on neuronal cells[14]. Furthermore, pro-inflammatory cytokines generated by activated microglia, such as IL-6 and TNF-α, have been reported to play important roles in neuronal injury and apoptosis[15]. Thus, more research has been done on the components that might inhibit microglial activation.
TET is known as a potent anti-inflammatory component. We performed studies on cultured microglial to address whether TET could suppress the activation of microglia. Rat-derived microglial cultures were established. The microglia pretreated with or without TET were stimulated by lipopolysaccharide (LPS) in vitro. Our results indicated that TET pretreatment inhibited NO release, O2– generation, as well as TNF-α and IL-6 production by microglia. These effects of TET may be attributed to its inhibitory effect on the NF-κB pathway during microglial activation.
Materials and Methods
Reagents Tetrandrine (purity >98%) was purchased from Huike Botanical Development (Shanxi, China). LPS from Escherichia coli (serotype 026:B6) was purchased from Sigma (St Louis, MO, USA). The enzyme-linked immunosorbent assay (ELISA) kits specific for rat TNF-α and IL-6 were purchased from R&D Systems (Minneapolis, MN, USA). Nucleotide oligomers were synthesized by Invitrogen (Carlsbad, CA, USA).
Microglia cultures and TET pretreatment Primary microglia culture was performed as described by Xiao et al[16]. Briefly, the brains were isolated from Sprague–Dawley rats (Shanghai SLAC Laboratory Animal, Shanghai, China) at postnatal d 1−3. After the meninges were carefully removed, the brains were minced mechanically. Dissociated cells were resuspended in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and seeded in 75-cm2 flasks at a density of 1×106 cells/mL. The cells were cultured in an incubator under 5% CO2 at 37 °C, and the medium was changed every 3 d. After 10 d, microglia growing on the surface of adherent astrocytes were removed by shaking for 4 h at 150 r/min, and the floating cells were collected and transferred to a 6-well plate at a density of 2×106 cells in 2 mL culture medium per well for 12 h before further treatment. The purity was 92%–95% for microglia as determined by anti-Mac-1 antibody (SeroTec, Oxford, UK) staining. In all the experiments, the cells were pretreated with TET (25 or 50 µmol/L) for 2 h before the addition of LPS (1 µg/mL). Control samples were pretreated with the culture medium only.
The study was performed according to the international, national, and institutional rules concerning animal experiments and biodiversity rights.
Measurement of nitrite The production of NO was determined by measuring nitrite, a stable derivation of NO, which reflects accumulated NO in the medium, using the Griess reagents (1% sulphanilamide, 5% phosphoric acid, and 0.1% naphthylethylenediamine). This assay was based on a diazotization reaction that was originally described by Griess in 1879[17]. Briefly, 100 µL of sample prepared from cells were mixed with 100 µL of Griess reagent in a 96-well flat plate. After 10 min, the plate was mounted in an automated microplate reader at 540 nm. The concentration of nitrite was determined by reference to a standard curve of sodium nitrite. Blank culture medium only was used as the control.
Measurement of O2– generation Superoxide radicals were generated by oxidation of NADH and analyzed by the reduction of nitroblue tetrazolium (NBT). In brief, the superoxide radicals were generated in 0.3 mL Tris-HCl buffer (16 mmol/L, pH 8.0) mixed with 0.1 mL NBT solution (50 mmol/L), 0.1 mL NADH solution (78 mmol/L), and an aliquot of the cell culture supernatant (20 µL). The reaction was initiated by adding 0.1 mL phenazine methosulphate solution (10 mmol/L) to the mixture. The reaction mixture was incubated at 25 °C for 5 min, and the absorbance at 550 nm in an automated microplate reader was measured against blank samples. L-Ascorbic acid was used as a negative control.
Analysis of cytokines TNF-α and IL-6 were quantified using a sandwich ELISA procedure. In brief, the wells of the microtiter plates were coated and stored overnight at room temperature with 100 µL phosphate-buffered saline (PBS) containing the appropriate dilutions of mouse antirat TNF-α antibody and mouse antirat IL-6 antibody. The coating was done at room temperature and kept overnight. The wells were then washed with 0.05% PBS (PBS–Tween-20) and blocked with 100 µL of 1% bovine serum albumin in PBS for 1 h at room temperature. After washing, 100 µL of the culture supernatants were added to the well in triplicates. After 2 h of incubation at room temperature, the wells were washed and incubated with 100 µL (0.4 mg/L) of biotinylated monoclonal antibodies (goat anti-rat TNF-α and goat anti-rat IL-6 antibodies) for 2 h at room temperature. After washing, 100 µL of streptavidin-horseradish peroxidase conjugates were added, and the plates were incubated for a further 45 min. After washing, 100 µL of substrate solution was then added to each well. The reaction was stopped after 20 min with a stopping solution. Optical density was measured at a wavelength of 450 nm and a reference wavelength of 540 nm. Density values were correlated linearly with the concentrations of cytokine standards (expressed in pg/mL).
Isolation of total RNA and RT–PCR Total RNA was isolated from cell pellets using the RNeasy mini kit (Qiagen, Hilden, Germany). Genomic DNA was removed from total RNA prior to cDNA synthesis by DNase digestion using the RNase-free DNase Set (Qiagen, Germany). RNA was stored at –80 °C. First-strand cDNA synthesis was performed for each RNA sample using the Sensiscript RT kit (Qiagen, Germany). Random hexamers were used to prime cDNA synthesis.
Real-time PCR The gene expression of iNOS was performed by real-time PCR using SYBR green master mix (Applied Biosystems, Foster City, CA, USA). The thermocycler conditions comprised an initial holding at 50 °C for 2 min, then 95 °C for 10 min. Reaction mixtures were cycled 40 times at 95 °C for 15 s and 60 °C for 60 s. Data were collected and quantitatively analyzed on an ABI Prism 7900 sequence detection system (Applied Biosystems, USA). The β-actin gene was used as an endogenous control to normalize differences in the amount of total RNA in each sample. All quantities were expressed in the number of folds relative to the expression of β-actin.
β-actin, sense: 5' TTCAACACCCCAGCCATGT 3' and antisense: 5' GTGGTACGACCAGAGGCATACA 3', and iNOS, sense: 5' CGGTTCACAGTCTTGGTGAAAG 3´ and antisense: 5' ACGCGGGAAGCCATGAC 3' .
Electrophoretic mobility shift assay After treatment with LPS (1 µg/mL) with or without TET for 24 h, microglia were collected. Buffer A (10 mmol/L HEPES, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/L phenylmethylsulfonyl fluoride [PMSF] and 1 mmol/L dithiothreitol [DTT]) was then added to the cells and incubated at 4 °C for 15 min, and 1.06 µL Nonidet P-40 (10%) was added. After centrifugation, the supernatant was removed and a high-salt solution (20 mmol/L HEPES, pH 7.9, 25% glycerol, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 1 mmol/L PMSF, and 1 mmol/L DTT) was added to the pellet. The resuspended pellet was incubated for 60 min at 4 °C, and then the supernatant was collected and used for the experiment. Protein concentrations were determined using a protein assay (Bio-Rad, Hercules, CA, USA). Synthetic double-stranded oligonucleotides for the consensus NF-κB binding sequence, 5' -AGTTGAGGGGACTTTCCCAGGC-3', were labeled with [γ-32P]dATP using T4 polynucleotide kinase (Promega, Madison, WI, USA). The nuclear extract from cultured cells was incubated with the labeled probe in Gel shift binding buffer (Promega, USA) at 4 °C for 30 min. DNA–protein complexes were resolved by electrophoresis in 4% polyacrylamide gels with 0.5×Tris-borate buffer at 4 °C and visualized by phorsphorimaging.
Statistical analysis All data are presented as mean±SD. Statistical comparisons between 2 different treatments were analyzed using the Student’s t-test. Differences among more than 2 groups were tested by one-way ANOVA. The levels of significance were set to α=0.05. All tests were 2 tailed.
Results
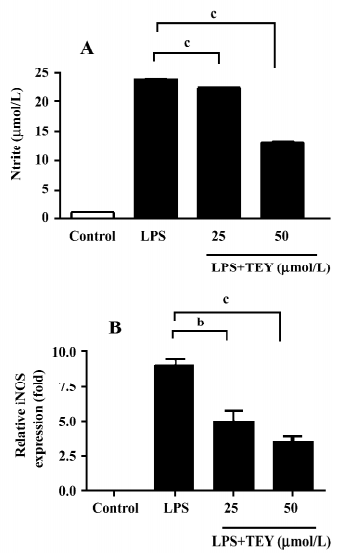
TET inhibited NO release and iNOS mRNA expression in LPS-activated microglia When rat primary microglial cells were stimulated with LPS in vitro, a strong induction of NO production was observed. The production of NO was determined by the measurement of nitrite, a stable product of NO, as an indicator to the accumulated NO in the medium (Figure 2A). Cultured microglia without LPS stimulation produced minimal NO in the supernatant. However, while stimulated by LPS (1 µg/mL for 24 h), microglia released significantly higher levels of NO from 1.11±0.06 to 23.89±0.25 µmol/L. The pretreatment of microglia with TET significantly reduced the elevation of NO release in a dose-dependent manner. The nitrite concentration was reduced to 22.57±0.10 (TET 25 µmol/L) and 13.34±0.28 (TET 50 µmol/L), respectively.
The cytotoxicity of TET was determined by LDH assay. There was no significant difference in LDH release between TET-treated and non-treated microglia. Therefore, the observed inhibition of NO was not due to the cytotoxicity of TET in microglia (data not shown).
In endotoxin-stimulated immune cells, iNOS is responsible for NO production. Therefore, we investigated whether TET suppressed iNOS expression induced by LPS. As shown in Figure 2B, TET downregulated the level of iNOS gene expression in LPS-activated microglia in a dose-dependent manner. These results suggest that TET regulates NO production in LPS-activated microglia via an iNOS-dependent pathway.
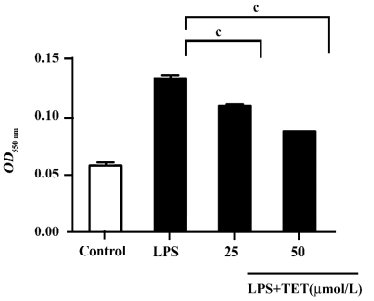
TET inhibited O2– production in LPS-activated microglia The LPS stimulation of microglia leads to the activation of NADPH oxidase and the production of reactive oxygen species (ROS), including O2–, which are ultimately responsible for the cellular oxidative damage[18]. We then tested whether TET could reduce the O2– generated by microglia stimulated with LPS. As shown in Figure 3, TET pretreatment inhibited LPS-stimulated O2– production in microglia in a dose-dependent manner. The inhibition reached a maximal value (35% reduction as compared to the control group) at a TET dose of 50 µmol/L.
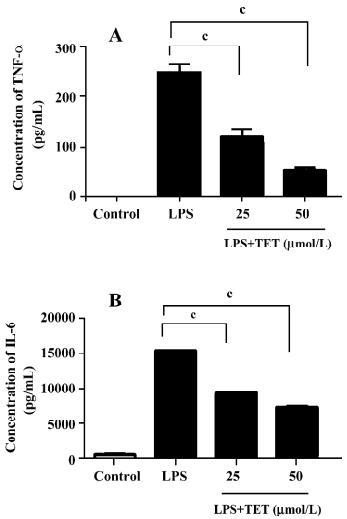
TET inhibited TNF-α and IL-6 release from LPS-activated microglia TNF-α and IL-6 generated by activated microglia play important roles in neurodegenerative diseases[19]. To examine the effect of TET on cytokine production by microglia, microglial cells were pretreated with TET for 2 h, followed by treatment with 1 µg/mL LPS for 24 h. The supernatants were then subjected to cytokine measurement. LPS stimulation markedly increased the TNF-α and IL-6 levels (Figure 4), which were significantly inhibited by TET pretreatment in a dose-dependent manner. Therefore, TET effectively suppressed the production of pro-inflammatory cytokines by activated microglia.
Effects of TET on the LPS-induced activation of NF-κB Because the NF-κB pathway is initially involved in LPS-induced microglial activation, we used the electrophoretic mobility shift assay (EMSA) to examine whether NF-κB activity in microglia was affected by TET treatment. Microglia were pretreated with TET (50 µmol/L) for 2 h and then stimulated with LPS (1 µg/mL) for 24 h. Nuclear extracts were prepared and EMSA was performed with a NF-κB consensus oligonucleotide. In microglia activated with LPS, nuclear NF-κB binding activity was enhanced (Figure 5A,5B). However, TET pretreatment dramatically abolished NF-κB specific binding induced by LPS, indicating that TET might downregulate the production of inflammatory mediators through the blockage of NF-κB activation.
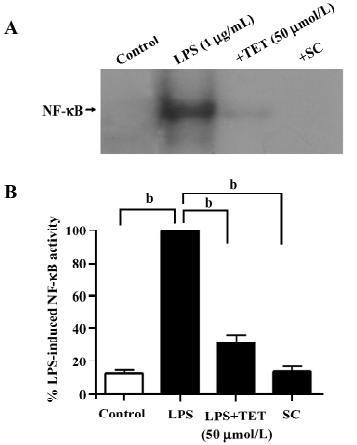
Discussion
To date, the role of microglia and whether they are neuroprotective or neurotoxic in the disease state of CNS remains controversial. Available evidence indicates that neurotrophic factors produced by activated microglia have a positive effect on the survival of neuronal cells. Nevertheless, microglial activation was also associated with neurodegenerative diseases. In microglial and neuronal coculture systems, microglial activation induced neuronal cell death by the release of NO, superoxide, and pro-inflammatory cytokines[20,21]. The inhibition of microglial activation could reduce neuronal cell death. In fact, studies of pathogenic events mediated by microglial activation in neurodegenerative diseases indicate that antineuroinflammatory agents have neuroprotective effects in these diseases. For example, many agents suppressed microglia-mediated neurotoxicity by inhibiting TNF-α, NO, or superoxide production, therefore exerting a neuroprotective effect[22–25].
TET exhibits immunosuppressive properties both in vitro and in vivo. In vitro, TET inhibited the production of NO in LPS-activated macrophages[26]. TET also inhibited the secretion of TNF-α by activated monocytes[8] and had an effect on cytokine production by activated T cells[27]. In a rat model of silicosis, TET effectively blocked the ability of quartz to stimulate oxidant release from pulmonary phagocytes[28]. Consistent with these studies, we demonstrated for the first time that TET showed significant suppressive effects on LPS-induced microglial activation.
Our data showed that TET inhibited iNOS mRNA expression and NO production in LPS-stimulated microglia. The high sensitivity of neurons to NO damage is partly due to the inhibition of respiration, rapid glutamate release, and subsequent excitotoxic damage. In our study, we also examined the amount of neuronal nitric oxide synthase (nNOS) and endothelial nitric oxide synthase (eNOS) mRNA expression with real-time PCR, but found them undetectable in microglia (data not shown).
We also found that TET dose dependently inhibited the generation of O2–. O2– is a short-lived free radical with important physiological functions. As an oxidant, it can in particular circumstances lead to the production of a variety of other stronger oxidants, such as hydrogen peroxide, the hydroxyl radical, hypochlorous acid, or peroxynitrite, all of which are cytotoxic. When appropriately stimulated, microglia can produce extracellular superoxide at very high rates[29]. In addition, NO can stimulate mitochondrial superoxide production and generate peroxynitrite within the mitochondria, which can irreversibly inhibit mitochondrial respiration or induce permeability transition[30]. The decline of O2– production as a result of TET treatment indicated a potential regulatory role of TET on ROS and the spread of neuronal damage during the development of inflammation and infection in the CNS. In previous studies, TET was also found to scavenge oxygen-derived free radicals directly in the cell-free hypoxanthine-xanthine oxidase system[31]. Therefore, whether the suppression of superoxide by TET resulted partially from the direct scavenging action needs further study.
Furthermore, our results illustrated that TET modulated the microglial production of TNF-α and IL-6, found to be secreted at a very early stage of CNS inflammation. TNF-α and IL-6 are elevated in most neurodegenerative diseases and there is evidence that they play critical roles in disease pathology. For example, TNF-α is a known potent inducer of cellular adhesion molecules in cerebrovascular endothelial cells[32] and astrocytes[33]. It can also induce chemokine expression in microglia[34,35] and astrocytes[35,36], thus promoting demyelination and oligodendrocyte injury[37]. IL-6 is also important for inflammatory response and is involved in reactive gliosis[38]. Therefore, augmentation of TNF-α and IL-6 leads to CNS inflammation and damage.
NF-κB is a major transcriptional factor for the induction of inflammation-related molecules. In resting cells, through masking the nuclear localization signal, NF-κB transcription factors are retained in the cytosol by inhibitory proteins, including inhibitory κBα (IκBα), IκBβ, IκBε, and IκBγ. After receiving a stimulatory signal, such as LPS, the IκBα inhibitory protein is phosphorylated by IκBα kinases, resulting in its ubiquitination and subsequent degradation by proteasome. These sequential yet highly regulated signal transduction events then cause nuclear translocation of NF-κB transcription factors[39]. NF-κB in the nucleus binds to the regulatory region in the gene promoter and is involved in the induction of iNOS and pro-inflammatory cytokines, including TNF-α and IL-6, in activated microglia. TET has been found to intervene with NF-κB activation and nuclear translocation in several cell lines[7,40]. TET could also prevent the degradation of IκBα and inhibit the nuclear translocation of p65 by blocking the activities of IκBα kinases a and b in human peripheral blood T cells[39]. Our findings in this study suggested that TET exerted its inhibitory effect on LPS-induced microglial activation by blocking the NF-κB-dependent pathway.
In conclusion, our results indicate for the first time that TET suppresses LPS-induced microglial activation and reduces the release of inflammatory mediators, including NO, O2–, and pro-inflammatory cytokines. Such suppressive effects are likely to be carried out through the inhibition of NF-κB activation. Together with previous experimental evidence that the suppression of microglial activation protects neuronal cells from various injuries, our findings raise the possibility of using TET in the treatment of neurodegenerative diseases.
References
- Ho LJ, Lai JH. Chinese herbs as immunomodulators and potential disease-modifying antirheumatic drugs in autoimmune disorders. Curr Drug Metab 2004;5:181-92.
- Xie QM, Tang HF, Chen JQ, Bian RL. Pharmacological actions of tetrandrine in inflammatory pulmonary diseases. Acta Pharmacol Sin 2002;23:1107-13.
- Lai JH. Immunomodulatory effects and mechanisms of plant alkaloid tetrandrine in autoimmune diseases. Acta Pharmacol Sin 2002;23:1093-101.
- Rao MR. Effects of tetrandrine on cardiac and vascular remodeling. Acta Pharmacol Sin 2002;23:1075-85.
- Shi X, Mao Y, Saffiotti U, Wang L, Rojanasakul Y, Leonard SS, et al. Antioxidant activity of tetrandrine and its inhibition of quartz-induced lipid peroxidation. J Toxicol Environ Health 1995;46:233-48.
- Chen F, Sun S, Kuhn DC, Lu Y, Gaydos LJ, Shi X, et al. Tetrandrine inhibits signal-induced NF-kappa B activation in rat alveolar macrophages. Biochem Biophys Res Commun 1997;231:99-102.
- Ho LJ, Chang DM, Lee TC, Chang ML, Lai JH. Plant alkaloid tetrandrine downregulates protein kinase C-dependent signaling pathway in T cells. Eur J Pharmacol 1999;367:389-98.
- Ferrante A, Seow WK, Rowan-Kelly B, Thong YH. Tetrandrine, a plant alkaloid, inhibits the production of tumour necrosis factor-alpha (cachectin) by human monocytes. Clin Exp Immunol 1990;80:232-5.
- Wang B, Yang L, Yan HL, Wang M, Xiao JG. Effect of tetrandrine on calcium-dependent tumour necrosis factor-alpha production in glia-neurone mixed cultures. Basic Clin Pharmacol Toxicol 2005;97:244-8.
- Gremo F, Sogos V, Ennas MG, Meloni A, Persichini T, Colasanti M, et al. Features and functions of human microglia cells. Adv Exp Med Biol 1997;429:79-97.
- Ryu JK, Shin WH, Kim J, Joe EH, Lee YB, Cho KG, et al. Trisialoganglioside GT1b induces in vivo degeneration of nigral dopaminergic neurons: role of microglia. Glia 2002;38:15-23.
- Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 1995;15:378-84.
- Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem 1997; 272: 29 390–7.
- Bal-Price A, Brown GC. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J Neurochem 2000;75:1455-64.
- Combs CK, Karlo JC, Kao SC, Landreth GE. Beta-amyloid stimulation of microglia and monocytes results in TNF-alpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 2001;21:1179-88.
- Xiao BG, Diab A, Zhu J, van der Meide P, Link H. Astrocytes induce hyporesponses of myelin basic protein-reactive T and B cell function. J Neuroimmunol 1998;89:113-21.
- Griess P. Bemerkungen zu der abhandlung der H.H.Weselsky und Benedikt “Ueber einige azoverbindungen”. Chem Ber 1879;12:426-8.
- McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 1997;17:2284-94.
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, . Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 2006; 281: 21 362–8.
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson’s disease. J Neurosci 2001;21:8447-55.
- Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 2003;27:325-55.
- Liu B, Jiang JW, Wilson BC, Du L, Yang SN, Wang JY, et al. Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther 2000;295:125-32.
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther 2000;293:607-17.
- Kowalski J, Labuzek K, Herman ZS. Flupentixol and trifluperidol reduce secretion of tumor necrosis factor-α and nitric oxide by rat microglial cells. Neurochem Int 2003;1289:1-6.
- Li R, Huang YG, Fang D, Le WD. (–)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J Neurosci Res 2004;78:723-31.
- Kondo Y, Takano F, Hojo H. Inhibitory effect of bisbenzylisoquinoline alkaloids on nitric oxide production in activated macrophages. Biochem Pharmacol 1993;46:1887-92.
- Lai JH, Ho LJ, Kwan CY, Chang DM, Lee TC. Plant alkaloid tetrandrine and its analog block CD28-costimulated activities of human peripheral blood T cells: potential immunosuppressants in transplantation immunology. Transplantation 1999;68:1383-92.
- Castranova V. Generation of oxygen radicals and mechanisms of injury prevention. Environ Health Perspect 1994.Suppl 10:65-8.
- Bal-Price A, Matthias A, Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem 2002;80:73-80.
- Riobo NA, Clementi E, Melani M, Boveris A, Cadenas E, Moncada S, et al. Nitric oxide inhibits mitochondrial NADH: ubiquinone reductase activity through peroxynitrite formation. Biochem J 2001;359:139-45.
- Seow WK, Ferrante A, Li SY, Thong YH. Antiphagocytic and antioxidant properties of plant alkaloid tetrandrine. Int Arch Allergy Appl Immunol 1988;85:404-9.
- Grau GE, Lou J. TNF in vascular pathology: the importance of platelet-endothelial interactions. Res Immunol 1993;144:355-63.
- Satoh J, Kastrukoff LF, Kim SU. Cytokine-induced expression of intercellular adhesion molecule-1 (ICAM-1) in cultured human oligodendrocytes and astrocytes. J Neuropathol Exp Neurol 1991;50:215-26.
- McManus CM, Brosnan CF, Berman JW. Cytokine induction of MIP-1α and MIP-1β in human fetal microglia. J Immunol 1998;160:1449-55.
- Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein-1 and other β-chemokines in murine glial cells. J Neuroimmunol 1995;60:143-50.
- Hurwitz AA, Lyman WD, Berman JW. Tumor necrosis factor α and transforming growth factor β upregulate astrocyte expression of monocyte chemoattractant protein-1. J Neuroimmunol 1995;57:193-8.
- Louis JC, Magal E, Takayama S, Varon S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 1993;259:689-92.
- Klein MA, Moller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6-deficient mice. Glia 1997;19:227-33.
- Ho LJ, Juan TY, Chao P, Wu WL, Chang DM, Chang SY, et al. Plant alkaloid tetrandrine downregulates IkappaBalpha kinases-IkappaBalpha-NF-kappaB signaling pathway in human peripheral blood T cell. Br J Pharmacol 2004;143:919-27.
- Chen F, Sun S, Kuhn DC, Lu Y, Gaydos LJ, Shi X, et al. Tetrandrine inhibits signal-induced NF-kappa B activation in rat alveolar macrophages. Biochem Biophys Res Commun 1997;231:99-102.