
Double-mismatched siRNAs enhance selective gene silencing of a mutant ALS-causing allele1
Introduction
Diseases caused by dominant, gain-of-function mutations develop in heterozygotes bearing 1 mutant and 1 wild-type copy of the gene. The best-known examples of this class are common neurodegenerative diseases, including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease). In these diseases, the exact pathways where the mutant proteins cause cell degeneration are not clear, but the origin of the cellular toxicity is known to be the mutant protein[1]. Thus, the design of compounds that selectively lower or eliminate the mutant protein is a key step in developing effective therapies.
Mutations in human CuZn superoxide dismutase (SOD) 1 cause motor neuron degeneration that leads to ALS because of some toxic property acquired by the mutant protein[2]. Although the molecular mechanism underlying this toxic property that triggers motor neuron degeneration is not fully understood, the artificial expression of mutant SOD1 in mice causes ALS. Nonetheless, the wild-type SOD1 gene is important for cells to function properly. SOD1 knockout mice demonstrate a variety of abnormalities, including reduced fertility[3], motor axonopathy[4], age-associated loss of cochlear hair cells[5], and neuromuscular junction synapses[6], and become vulnerable to noxious assaults, such as axonal injury[7], ischemia[8,9], hemolysate exposure[10], and irradiation[11]. Given the functional importance of the wild-type allele and the unwanted effect of the mutant protein, selectively blocking the expression of the mutant allele while keeping the expression of the wild-type protein intact would be an ideal therapy for the fraction of ALS caused by mutant SOD1.
Sequence-specific gene silencing has been achieved in eukaryotes by RNA interference (RNAi)[12–14]. Subsequent studies of the RNAi pathway[15–18] have been extended to cultured mammalian cells[19,20]. In this approach, 21 nt, double-stranded synthetic siRNA are used to destroy a target mRNA containing the corresponding siRNA sequence. This newly-developed technology holds a potential to cure diseases caused by dominant, gain-of-function gene mutations, such as ALS.
Previously, we showed that RNAi could selectively inhibit the expression of the mutant SOD1 genes that cause ALS[21]. In this study, we systematically tested a siRNA design strategy in converting a naturally symmetric siRNA to a favorable asymmetric siRNA that targets an ALS-causing SOD1 mutant allele in human cells. We demonstrate that this strategy can not only successfully achieve the conversion of an siRNA that is originally favored to the ant-sense of the mutant allele to the one that is favored to the sense strand of the gene, but also enhances the selective silencing of the target gene.
Materials and methods
siRNA preparation Chemically-synthesized single-strand RNA were purchased from Dharmacon Research (Chicago, USA), deprotected according to the manufacturer’s instructions, and annealed as described previously[21].
Cell culture and transfection Human embryonic kidney 293 cells were cultured in Dulbeccos’s Modified Eagle’s Medium (Invitrogen, Carlsbad, USA) supplemented with 10% fetal bovine serum plus 100 U/mL penicillin and 100 µg/mL streptomycin. Twenty-four hours before the experiments, the cells were detached with trypsin-EDTA (0.05% trypsin, 0.53 mmol/L EDTA·4Na) at 70%–90% confluency and transferred into 96-well plates at approximately 30% cell density. Transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) following the manufacturer’s instructions on the following day. The concentrations of the constructs and siRNA used in the transfection were 2.0 µg/mL firefly luciferase (pGL2 control vector, Promega, San Luis Obispo, USA) with target sequence inserted into the 3’ untranslated region (UTR), 0.1 µg/mL of Renilla luciferase vector (pRL-TK, Promega, San Luis Obispo, USA), and 2 nmol/L siRNA. The siRNA concentrations used for the dose-effect study (Figure 1) were 0.002–31.25 nmol/L. Data are presented as mean±SD (n=4).
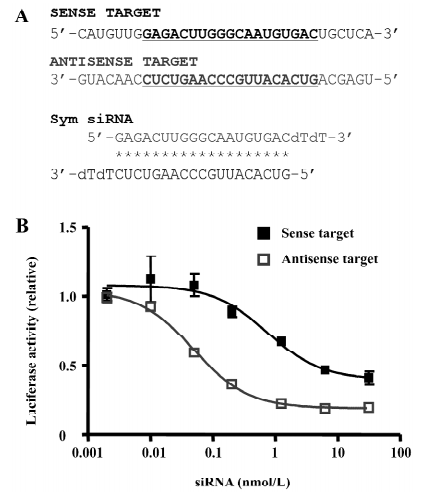
Dual luciferase assay The dual luciferase system (Promega, San Luis Obispo, USA) was used with modifications to quantify RNAi efficiency in the cell culture. Two restriction sites (Nde l and Spe I) were first engineered into the 3' UTR of the firefly luciferase vector followed by the insertion of a 39 nt fragment of the SOD1 gene (sense strand5’AGGCATGTTGGAGACTTGGGCAATGTGACTGCTGA-CAAA-3', anti-sense strand 5'-TTTGTCAGCAGTCACA-TTGCCCAAGTCTCCAACATGCCT-3') between the Nde I and Spe I sites. The modified firefly luciferase vector with either the partial SOD1 sense (sense target) or antisense sequence (antisense target) was cotransfected with the Renilla luciferase vector plus siRNA into HEK293 cells in quadruplicate. Twenty-four hours after the transfection, the cells were lysed in 96-well plates with 20 µL passive lysis buffer (Promega, San Luis Obispo, USA). In total, 10 µL lysate was transferred into a well in a Microlite strip (Thermo Labsystems, Waltham, USA), and the luminescence intensity was measured with a Veritas microplate luminometer (Turner Biosystems, Sunnyvale USA). The relative ratio of the firefly/Renilla luciferase was used for calculating the RNAi efficacy. All measures were normalized to the luciferase vectors only with vectors plus siRNA against green florescent protein (GFP) mRNA as an irrelevant control. The results were plotted using mean±SD (n=4; Figures 2,3,4).
Results
The siRNA that we started with was an siRNA targeting the ALS-causing mutant G85R SOD1 gene, which has been previously shown to be inefficient in silencing the mutant allele and has almost no effect on the gene expression of wild-type SOD1[21]. This natural siRNA has the exact same base pairing at the first 4 positions of both ends, therefore is symmetric (Figure 1A). To quantify the effects of the siRNA, we transfected the sense or antisense targets with different doses of the siRNA. This original symmetric siRNA simultaneously silenced the sense and antisense targets. However, the maximal silencing effect (relative) for the antisense target was 81%, with the IC50 (50% inhibitory concentration) at 0.1 nmol/L, while the efficiency for the cleavage of the sense target, the ALS-causing sequence, was only 60% with the IC50 at 5 nmol/L (Figure 1B).
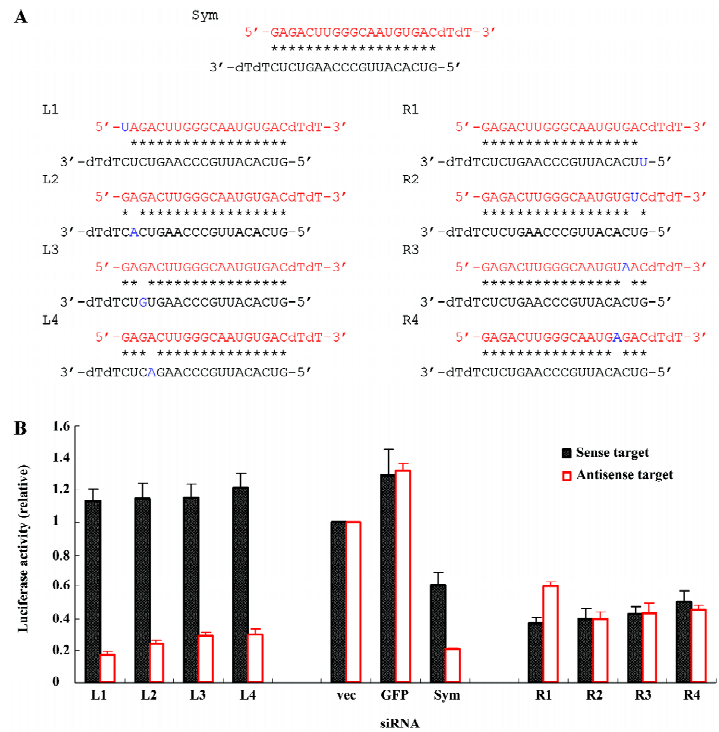
To convert the naturally symmetric siRNA that is favored to the antisense stand of ALS-causing gene to the effective siRNA that targets the disease-causing sequence itself, we weakened the base pairing at the 5' of the antisense strand of the siRNA (right end, R) by placing a mismatch at that end (R1–R4; Figure 2A). Although the symmetric siRNA naturally favored the antisense target (Figure 1B), it was converted to favor the sense target after weakening the base pairing at the 5' of the antisense strand of the siRNA (R1–R4; Figure 2B). The mismatch at position 1 (R1) completely reversed the strand preference compared to the original symmetric siRNA, while the efficacies of mismatches from positions 2 to 4 (R2–R4) decreased as the position moving towards the middle of the strand. In contrast, the weakening of the base pairing at the 5' of the sense strand of the siRNA (left end, L) accentuated the preference of silencing the antisense target (L1–L4; Figure 2B). All 4 siRNAs with a mismatch from positions 1 to 4 (L1–L4; Figure 2A) abolished the silencing of the sense target. Meanwhile, they effectively silenced the antisense target and the siRNA with a mismatch at position 1 (L1) exhibited the maximal efficacy.
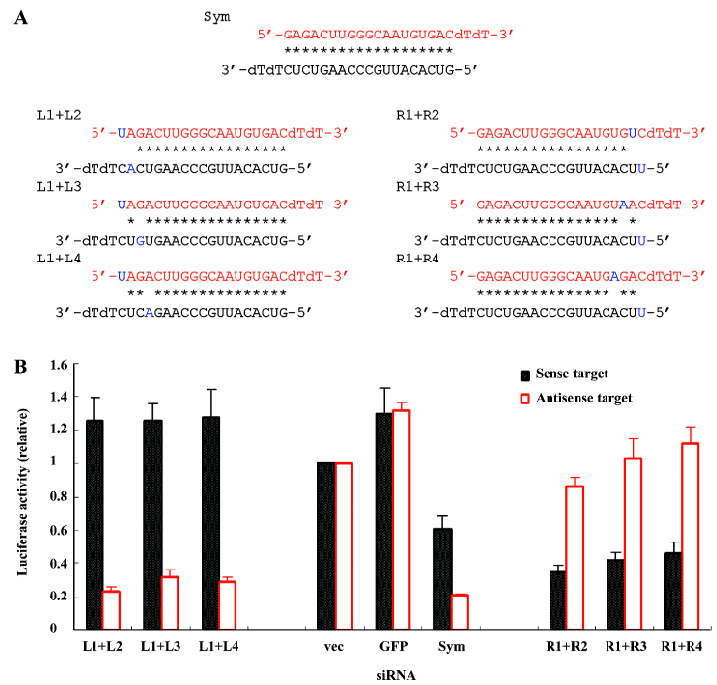
To further weaken the base pairing, we placed 2 mismatches in the symmetric siRNA (Figure 3A). When the mismatches were placed at the 5' end of the sense strand of the siRNA, the target preference of this double-mismatched siRNA was only slightly increased (L1+L2, L1+L3, and L1+L4; Figure 3B) compared to one of the single-mismatched siRNA, mainly because placing the 1 mismatch at this end of the siRNA almost maximized the siRNA asymmetry already (L1, L2, L3, and L4; Figure 2B). However, if the mismatches were inserted into the 5' end of the antisense strand of the siRNA where placing 1 mismatch did not completely convert the siRNA from symmetry to asymmetry; the modified siRNA noticeably increased the efficacy of the sense target cleavage and dramatically decreased the inhibition of the antisense target (R1+R2, R1+R3, and R1+R4; Figure 3B). Thus, double-mismatched siRNA in general improved the preference and efficacy of target silencing compared to single-mismatched siRNA.
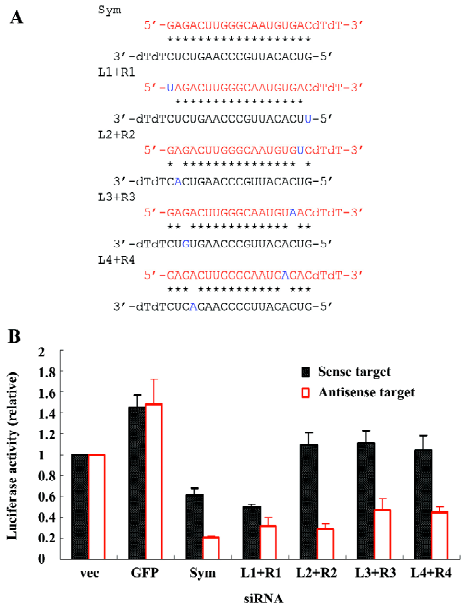
Finally, we tested the silencing efficacies of siRNAs with mismatches at both ends by placing 1 of the 2 mismatches at each end of the siRNA at corresponding positions (L1+R1, L2+R2, L3+R3, L4+R4; Figure 4A). These siRNAs obtained a symmetric mismatched structure and exhibited a similar pattern of target silencing to the one of the original symmetric siRNA that prefers the inhibition of the anti-sense target expression, although the extent of target cleavage was slightly different among the modified symmetric siRNAs with double-mismatches (Figure 4B).
Discussion
RNAi has become a powerful tool in reverse genetics for the investigation of gene functions in recent years. More importantly, it has been increasingly applied as therapeutic strategies in cells, animals, and potentially in humans[22]. The silencing of mutant genes that cause ALS is one of the leading applications of gene therapy by RNAi. The design of effective siRNAs is the key for the success of RNAi therapy.
Poor siRNA efficacy caused by the inaccessibility of the target region[23,24] and the unfavorable strand asymmetry of the siRNA[25,26] could limit the therapeutic use of RNAi. According to the asymmetric rule of RNAi, only 1 strand of each siRNA will be loaded into the RNA-induced silencing complex (RISC) and cleave its target mRNA. The other strand will be degraded without executing RNAi. The thermodynamic stability of base pairing at the 2 ends of the siRNA determines which strand will function. The strand with a less stable 5' base pairing than its 3' base pairing is more likely to enter the RISC. For a symmetric siRNA, the stability of base pairing at the 2 ends is the same so that both strands enter the RISC equally and mediate RNAi with similar potencies. Apparently, an unfavorable asymmetric siRNA that has stability of its end base pairing favoring the sense strand to enter the RISC will silence the complementary strand of the target mRNA (antisense target) and therefore has poor RNAi efficacy on the intended target (sense target).
Previous designs of siRNA have focused on searching naturally asymmetric siRNAs[7,27]. This approach may be limited in situations, such as ALS-causing mutant SOD1 genes, where the target region is confined by the location of a mutation and within this confined region no favorable asymmetric siRNAs can be obtained. Alternatively, weakening base pairing by the incorporation of mismatches at the 5' of the antisense strand of an siRNA can create strand asymmetry favoring the silencing of an intended gene[22,26].
In the present study, we tested the strategy to covert the symmetric siRNA to the asymmetric one to increase the capacity of siRNA in the selective silencing of target genes. Consistent with previous findings[26], our study demonstrated that a single mismatch at one end of the siRNA targeting a mutant SOD1 gene (G85R) successfully disrupted its original symmetry that led to the non-selective cleavage of both sense and antisense targets and turned the siRNA into asymmetric siRNA, thus selectively silencing either sense or antisense targets (Figure 2). To further modify the asymmetric design for generating more effective siRNAs, we systematically modified the symmetric siRNA with double mismatches. The new siRNAs with double mismatches at one end demonstrated an increased asymmetry compared to their single-mismatched counterparts (Figure 3). In addition, double mismatched siRNAs that had 1 mismatch at each end of the original symmetric siRNA remained in symmetry as expected (Figure 4). These results suggest the effectiveness of converting a symmetric siRNA to an asymmetric one by introducing mismatches into its structure, and the superiority of double-mismatched siRNA to single-mismatched siRNA in producing effective gene silencing due to the disruption of siRNA symmetry. More importantly, by placing double mismatches at the 5’ end of the antisense of the siRNA (R1+R2, R1+R3, R1+R4; Figure 3), the modified siRNA enhanced the silencing of mutant SOD1 gene expression while abolishing the potential harmful cleavage of the antisense sequence of this mutant allele. Therefore, we can achieve a gene silencing of the ALS-causing mutant SOD1 and avoid unwanted blockage of other functionally important genes with partial homology to the antisense sequence of the mutant gene (off-target effect). Although we can not completely exclude the possibility that the decrease of target cleavage might partially result from a single nucleotide mismatch between either the sense or antisense strand of siRNA and its target, it is unlikely to be a major contributor of the pattern change in the efficacy or specificity of RNAi observed in our experiments because the majority of these mismatches falls into the first 3 positions at the end of siRNA in which a single nucleotide mismatch has demonstrated inability of substantially affecting the selectivity of siRNA[28]. Overall, the findings from this study not only confirm the prediction of asymmetric rule in the function of siRNA, but also provide convincing evidence for a better strategy in the design of effective siRNAs for gene therapy.
ALS is a lethal neurodegenerative disease without a cure. The establishment of a connection between mutations in the SOD1 gene and familiar ALS has provided an entry point to elucidate the underlying mechanism and find an effective treatment for the disease. Our previous study showed a feasibility of allele-specific silencing of ALS-causing mutant alleles by RNAi[21]. Here, we further demonstrated an improved strategy in the design of effective siRNAs for the gene therapy for the ALS type that is caused by a mutant SOD1 (G85R). Since there are more than 100 mutations in SOD1 that can lead to ALS, the success of gene silencing by RNAi in these mutant genes may prevent many new cases from occurring. Nevertheless, more tests need to be performed to ensure the generalizability of this strategy to different SOD1 mutant alleles and to other diseases caused by dominant, gain-of-function gene mutations.
Acknowledgements
We thank Dr Zuo-shang XU, Dr Dianne SCHWARZ, and Dr Phillip ZAMORE for advice and sharing reagents. This work was supported by grants from the ALSAssociation, the NIH/NINDS (N
References
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegen-erative disease. Science 2002;296:1991-5.
- Cleveland DW, Rothstein JD. From charcot to lou gehrig: Deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001;2:806-19.
- Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology 1998;139:4008-11.
- Shefner JM, Reaume AG, Flood DG, Scott RW, Kowall NW, Ferrante RJ, et al. Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy. Neurology 1999;53:1239-46.
- McFadden SL, Ding D, Salvi R. Anatomical, metabolic and genetic aspects of age-related hearing loss in mice. Audiology 2001;40:313-21.
- Flood DG, Reaume AG, Gruner JA, Hoffman EK, Hirsch JD, Lin YG, et al. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am J Pathol 1999;155:663-72.
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol 2004;22:326-30.
- Kawase M, Murakami K, Fujimura M, Morita-Fujimura Y, Gasche Y, Kondo T, et al. Exacerbation of delayed cell injury after transient global ischemia in mutant mice with CuZn superoxide dismutase deficiency. Stroke 1999;30:1962-8.
- Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, et al. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 1997;17:4180-9.
- Matz PG, Copin JC, Chan PH. Cell death after exposure to subarachnoid hemolysate correlates inversely with expression of CuZn-superoxide dismutase. Stroke 2000;31:2450-9.
- Behndig A, Karlsson K, Reaume AG, Sentman ML, Marklund SL. In vitro photochemical cataract in mice lacking copper-zinc superoxide dismutase. Free Radic Biol Med 2001;31:738-44.
- Hutvagner G, Zamore PD. RNAi: Nature abhors a double-strand. Curr Opin Genet Dev 2002;12:225-32.
- Hannon GJ. RNA interference. Nature 2002;418:244-51.
- McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 2002;3:737-47.
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000;101:25-33.
- Hamilton AJ, Baulcombe DC. A species of small anti-sense RNA in posttranscriptional gene silencing in plants. Science 1999;286:950-2.
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature 2000;404:293-6.
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev 2001;15:188-200.
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 2001;98:9742-7.
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494-8.
- Ding H, Schwarz DS, Keene A. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003;2:209-17.
- Uprichard SL. The therapeutic potential of RNA interference. FEBS Lett 2005;579:5996-6007.
- Heale BS, Soifer HS, Bowers C, Rossi JJ. siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res 2005;33:e30.
- Brown KM, Chu CY, Rana TM. Target accessibility dictates the potency of human RISC. Nat Struct Mol Biol 2005;12:469-70.
- Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003;115:209-16.
- Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003;115:199-208.
- Chalk AM, Warfinge RE, Georgii-Hemming P, Sonnhammer EL. siRNAdb: A database of siRNA sequences. Nucleic Acids Res 2005;33:D131-4.
- Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, et al. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet 2006;2:e140.