
α1B-Adrenoceptors mediate adrenergically-induced renal vasoconstrictions in rats with renal impairment
Introduction
The sympathetic nervous system plays an important role in the control of renal hemodynamics[1,2]. In renal vasculature, the catecholamines released from the nerve terminals activate G protein-coupled cell surface adrenoceptors present on the cell surface and cause contraction[2,3]. Three subtypes of α1-adrenoceptor subtypes, namely α1A, α1B, and α1D, have been documented based on pharmacological and cloning studies[4–7]. These subtypes are closely related to each other in terms of amino acid sequence, ligand affinity, and also with respect to irreversible blockade by certain alkylating agents[8]. Several in vivo and in vitro studies have revealed that, functionally, α1-adrenoceptors predominate in the renal vasculature[9–11]. In rat renal vasculature, all of the 3 subtypes of α1-adrenoceptors have been shown to mediate catecholamine-induced constriction[2,9–11] along with a dominant role of the α1A-subtype[10,11]. Some reports have also shown the functional involvement of other subtypes, such as α1D-adrenoceptors, in renal vasculature[2,12–14].
The α1B-adrenoceptors are also involved in mediating vasoconstrictions as observed in in vitro studies with the mesenteric resistance artery and tail artery of rats[15,16]. This subtype is also present in the renal resistance vessel and receptor binding. The RNase protection assay revealed the expression of α1B-adrenoceptors in the renal microvessels of Wistar Kyoto and spontaneously hypertensive rats. However, there is no report on the functional contribution of this subtype in modulating adrenergically-induced renal vasoconstriction except reports on its minor involvement. A minor functional involvement of α1B-adrenoceptors in mediating renal vasoconstriction is reported in rats with some altered physiological or pathological states. In diabetes, hypertensive diabetes, heart failure, and renal failure, a minor involvement of this receptor subtype has been reported in mediating renal adrenergic responsiveness in rats[4,17,18]. Some pathological states are indeed found to influence the functional contribution of α1-adrenoceptors, including the α1B-subtype, in rat renal vasculature[14,18,19]. However, there is still an apparent paucity of information on the functional involvement of α1B-adrenoceptors in modulating renal vasoconstrictions that could strengthen this view. It is particularly true in terms of further study of this interesting observation in some other pathological states.
The present study investigated the influence of some pathological states with renal impairment on the functional presence of α1B-adrenoceptors in terms of its selective affinity for chloroethylclonidine. The results of this study will provide further evidence on the pathological state-dependent involvement of α1B-adrenoceptors in modulating adrenergically-induced renal vascular tone in rats.
Methods and materials
Animals Male Wistar Kyoto and spontaneously hypertensive rats with (body weight range: 250–300 g, 267±8.3 g) were supplied with commercial rat chow and water ad libitum. They were maintained in the Animal Care Facility, Universiti Sains Malaysia, Penang, Malaysia. Animal handling and all animal procedures were approved by the Animal Ethics Committee, Universiti Sains Malaysia. The animals were randomly divided into 6 groups. Groups 1 and 2 (n=5–9 rats in each group) were normal and renal failure Wistar Kyoto rats, groups 3 and 4 (n=5–9 rats in each group) were normal and renal failure spontaneously hypertensive rats, and groups 5 and 6 (n=5–9 rats in each group) were rats with experimental early diabetic nephropathy and the non-diabetic nephropathy control group, respectively.
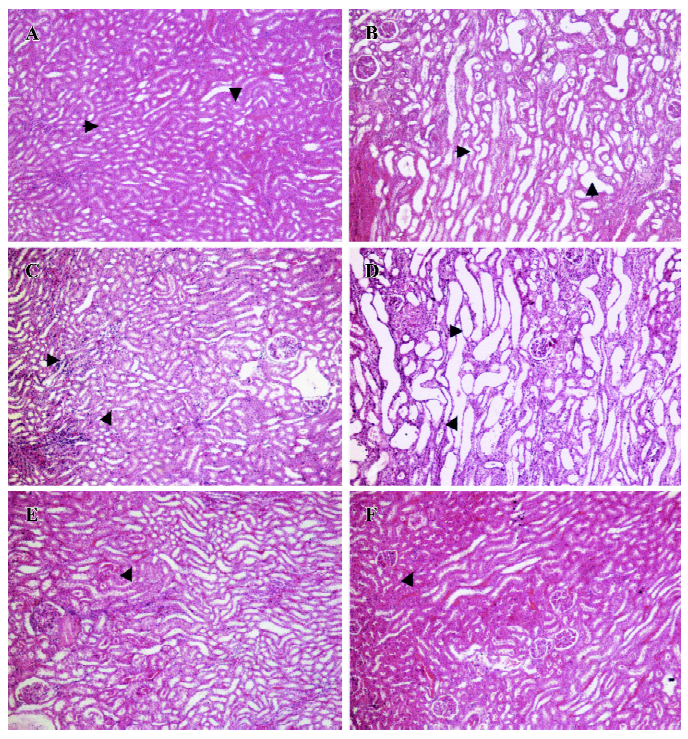
Induction of renal failure and physiological data collection The animals were caged individually in custom-built stainless steel metabolic cages and acclimatized for at least 3 d before the induction of renal failure with cisplatin. Baseline physiological data (body weight, 24 h water intake, and urine output) were recorded. The animals fasted for at least 12 h and on the following day received a single intraperitoneal injection of cisplatin (55 mg/kg)[20–22]. Further physiological data were collected on every alternate day until the animals were used in the acute renal hemodynamic study on d 7. Tail vein blood samples were collected on d 0 and 7, and plasma were separated and frozen (-70 oC) until analyzed for creatinine and sodium using spectrophotometry and flame photometry, respectively. The kidney index was calculated as 100×kidney weight/body weight[23–25]. The kidney tissues were preserved in 10% formalin for the histological examinations using conventional hematoxylin and eosin staining followed by the analysis of micrographs (Leica image analyzing system, London, UK). Apart from the examinations of the kidney index and histological study of the kidney, renal failure and impairment of renal functions in these rats were assessed from plasma creatinine, creatinine clearance, fractional excretion of sodium, and the glomerular filtration rate.
Preparation of experimental early diabetic nephropathy rats and physiological data collection The rats with experimental early diabetic nephropathy were developed using spontaneously hypertensive rats by a slightly modified method described earlier by several researchers[25,26–28]. In this approach, the spontaneously hypertensive rats were treated with a single intraperitoneal injection (55 mg/kg) of streptozotocin after 12–16 h of fasting. On d 3 of post-streptozotocin injection fasting (
Apart from elevated blood glucose, other physiological changes, such as polyuria and a reduction in the body weight, were also considered in selecting the diabetic animals. The diabetic rats were kept for 28 d for weekly physiological data collection and also for the development of changes in renal functional parameters, such as creatinine clearance, fractional excretion of sodium, glomerular filtration rate, and the urinary albumin excretion rate. Creatinine and sodium in the plasma and urine were measured using spectrophotometry and flame photometry, respectively, and urinary albumin was measured using ELISA[31]. Finally, on d 29 the rats were used in the acute study for renal hemodynamic data collection. After the hemodynamic study, the kidney tissues were collected, the weight of the kidneys was recorded for the determination of the kidney index (kidney index=100×kidney weight/body weight), and the kidneys were then preserved in 10% formalin for the histological examinations.
Hemodynamic study
Surgical preparation of the animal The overnight (≥12 h)-fasted (with water ad libitum) rats were anaesthetized with 60 mg/kg (ip) sodium pentobarbitone (Nembutal, CEVA Sante Animale, Libourne, France). After a tracheostomy with endotracheal cannula (PP240, Protex, Kent, England) the carotid artery was cannulated (PE 50, Protex, England) and connected to a pressure transducer (P23 ID Gould, Statham Instruments, Oakland, CA, USA) coupled to a computerized data acquisition system (PowerLab, AD-Instruments, Sydney, NSW, Australia) for the continuous measurement of the mean arterial blood pressure. The left jugular vein was cannulated (PE 50, Protex, England) to permit the infusion of maintenance doses of anesthesia.
The left kidney was exposed using a midline abdominal incision. The renal artery was carefully cleared and fitted with an electromagnetic flow probe (EP 100 series, Carolina Medical Instruments, King, North Carolina, USA) that was connected to an electromagnetic flow meter (Carolina Medical Instruments, USA) for the continuous measurement of renal blood flow. A cannula (PE 50, Protex, England) was inserted via the iliac artery so that its beveled tip lay close to the entrance of the renal artery to enable the exogenous administration of adrenergic agonists and antagonists[10,11]. The cannula was kept patent by the continuous infusion of saline at a rate of 6 mL·kg-1·h-1. The renal nerves passing from the coeliac and aortico-renal ganglia to the kidney were isolated and carefully dissected for a short length and placed on fine bipolar stainless steel wire electrodes. The functionality of the renal nerves was tested by stimulating (Grass S 48 Stimulator, Grass Instruments, Quincy, MA, USA) them at 15 V, 0.2 ms, and 1–10 Hz for 30 s to observe whether blanching of the kidney occurred. At the end of the experiment, the animals were euthanized by an overdose of anesthetic (sodium pentobarbitone; Nembutal, CEVA Sante Animale, France), and an autopsy was done followed by the disposal of the animal carcasses in accordance with the guidelines of the Animal Ethics Committee of the Universiti Sains Malaysia.
Experimental protocol The experiment was comprised of 3 phases. In the first phase, saline was infused continuously and intrarenally during which the renal nerves were stimulated at increasing frequencies of 1, 2, 4, 6, 8, and 10 Hz and then in the reverse order. Subsequently, graded bolus doses of noradrenaline (25, 50,100, and 200 ng; Sanofi Winthrop, Guildford, UK), phenylephrine (0.25, 0.5, 1, and 2 μg; Knoll, Nottingham, UK), and methoxamine (1, 2, 3, and 4 μg; Wellcome, London, UK) were administered in ascending and then descending doses. After the first phase, a close intrarenal administration of a bolus dose (5 μg/kg) of chloroethylclonidine (Sigma, St Louis, MO, USA) was given followed by a continuous infusion of 1.25 μg·kg-1·h-1. Twenty minutes later, the second set of vasoconstriction experiments were carried out. In the last phase, a bolus dose of 10 μg/kg chloroethylclonidine plus a continuous infusion of 2.5 μg·kg-1·h-1 was administered. Twenty minutes latter the last set of vasoconstriction experiments were carried out as described earlier[10,11].
Renal vasoconstrictor responses The vasoconstrictor responses were recorded as the percentage changes of renal blood flow in relation to the baseline values during graded frequencies of renal nerve stimulation and graded doses of agonist administered. The responses were recorded using a computerized data acquisition system (PowerLab, ADInstruments, Australia).
Adrenergic agonist and antagonists Chloroethyl-clonidine (N-β-chloroethyl-N-methylamino-methyl-clonidine), a selective antagonist of α1B-adrenoceptors and the most widely used agent in characterizing α1B-adreno-ceptors, was used in this study for characterizing the functional involvement of α1B-adrenoceptors in mediating adrenergically-induced renal vasoconstriction, and can differentiate between α1A- and α1B-adrenoceptor subtypes[32]. Regarding the α1-adrenoceptors, the effects of chloroethyl-clonidine have been related to the alkylation and inactivation of the α1-adrenoceptor subtypes as irreversible antagonist in the order of sensitivity of the α1B ≥α1D>>α1A-adreno-ceptor subtypes[14,33]. Chloroethylclonidine was prepared in saline as recommended by the manufacturer and kept as aliquots of frozen stock and diluted prior to use.
The adrenergic agonists used were noradrenaline, phenylephrine, and methoxamine. Noradrenaline is a mixed agonist that acts on both the α1 and α2-adrenoceptors; phenylephrine is a non-selective agonist of α1-adrenoceptors with an ability to activate α1A-, α1B-, and α1D-adrenoceptor subtypes[14]; and methoxamine is a relatively selective α1A-adrenoceptor-subtype agonist[13,14], but may activate α1D-adrenoceptors as it does not exhibit selectivity between α1A- and α1D-adrenoceptors[13]. Noradrenaline, phenylephrine, and methoxamine were prepared fresh in saline (150 mmol/L NaCl) from frozen stocks daily prior to use.
Statistical analysis The renal blood flow responses caused by renal nerve stimulation, noradrenaline, phenylephrine, and methoxamine were taken as the average values caused by each dose/frequency of adrenergic stimuli administered and applied in ascending and descending orders. The overall mean response for each dose or frequency was taken as the average value of vasoconstrictor responses (drop in renal blood flow) obtained at each level of frequency for renal nerve stimulation and each dose of the adrenergic agonists used. The data on the drop of renal blood flow were expressed as the percentage drop on the renal blood flow in relation to the basal values of renal blood flow calculated at the beginning of the administration of each stimulus (renal nerve stimulation and adrenergic agonists) used. All data were expressed as mean percentage change±SEM of renal vasoconstrictor responses elicited by all the frequencies (renal nerve stimulation) and all the doses (adrenergic agonists) and were compared between the phases (saline, low dose of chloroethylclonidine, and high dose of chloroethylclonidine-treated phases). In the renal vasoconstriction experiments, two-way ANOVA was used for the statistical analysis. For the analysis of the physiological and other data, one-way ANOVA was used followed by the Bonferroni post-hoc test (Superanova, Abacus, Barkley CA, USA). The differences between the means were considered significant at the 5% level. All physiological and biochemical data (body weight, 24 h water intake, 24 h urine output, plasma sodium, urinary sodium, fractional excretion of sodium, creatinine clearance, albumin excretion, glomerular filtration rate, and kidney index) were analyzed using one-way ANOVA followed by the Bonferroni post-hoc test[8,10,11,14].
Results
General observations Renal failure was identified by increased plasma creatinine, reduced creatinine clearance, increased fractional excretion of sodium, reduced glomerular filtration rate, and diuresis in the cisplatin-treated renal failure rats (all P<0.01) as compared to the non-cisplatin-treated non-renal failure animals (Table 1). Moreover, there was a markedly increased (P<0.01) kidney index (percentage of kidney weight to body weight) and pronounced renal tubular damage in the renal failure as compared to the non-renal failure rats (Table 1; Figure 1). In the experimental early diabetic nephropathy rats, along with marked hyperglycemia, there was increased serum creatinine, creatinine clearance, urinary albumin excretion, glomerular filtration rate (all P<0.01), moderately increased fractional excretion of sodium (P>0.05), and an increased kidney index (P<0.01). Unlike the renal failure rats, there was no adverse tubular structural change in these rats as compared to the non-diabetic nephropathy control rats (Table 1; Figure 1). The baseline mean arterial blood pressure and renal blood flow of all the experimental groups are presented in Table 2. In the renal failure rats, renal blood flow was significantly lower compared to the non-renal failure rats (P<0.01). In the experimental early diabetic nephropathy rats, renal blood flow was greater compared to the non-diabetic nephropathy rats. However, this change in renal blood flow did not attain statistical significance (P>0.05). In addition, the baseline mean arterial blood pressure of these rats (renal failure and experimental early diabetic nephropathy) was either lower or unaltered (P>0.05) in the diseased rats when compared with the non-renal failure or non-diabetic nephropathy rats.
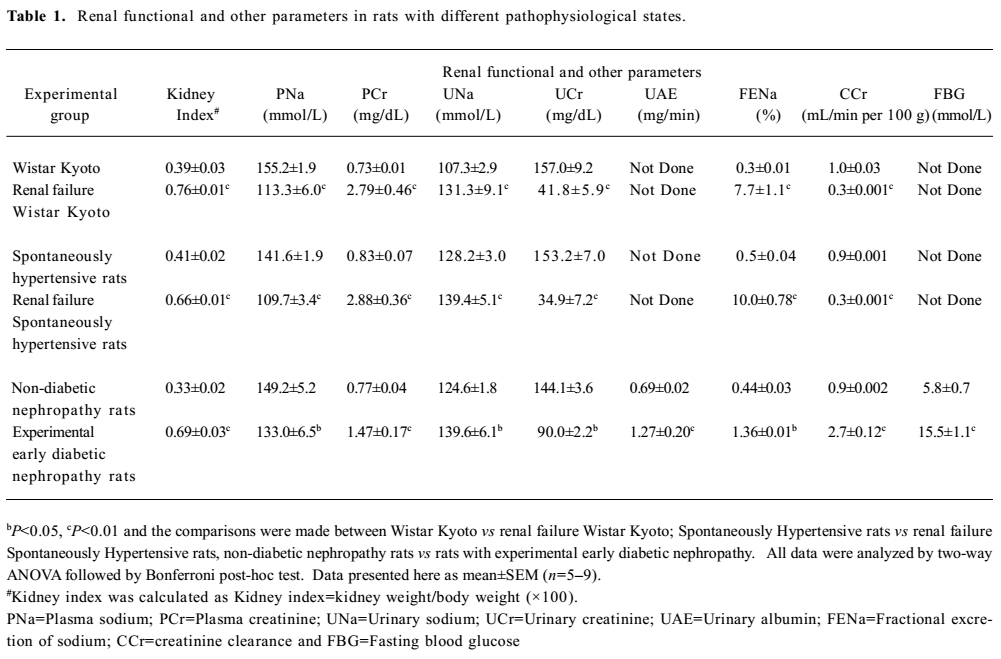
Full table
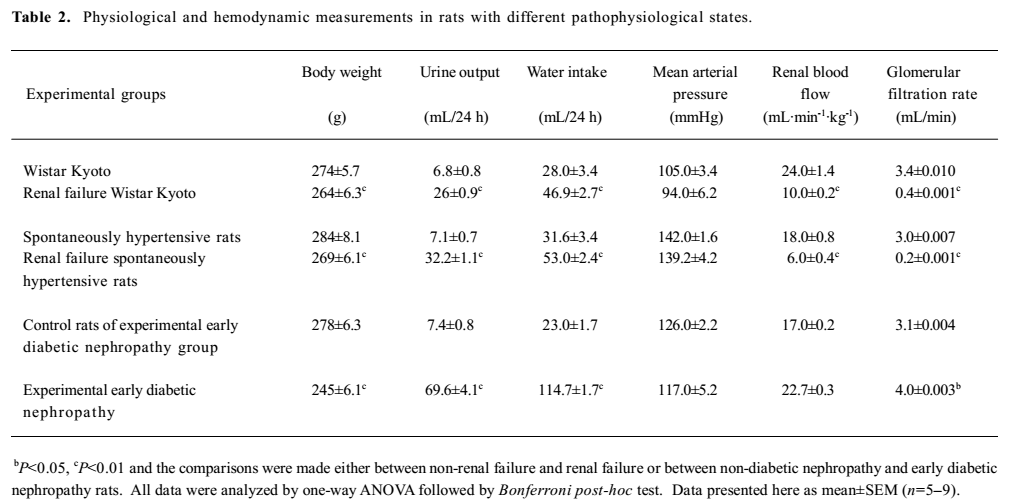
Full table
Renal vasoconstrictor responses
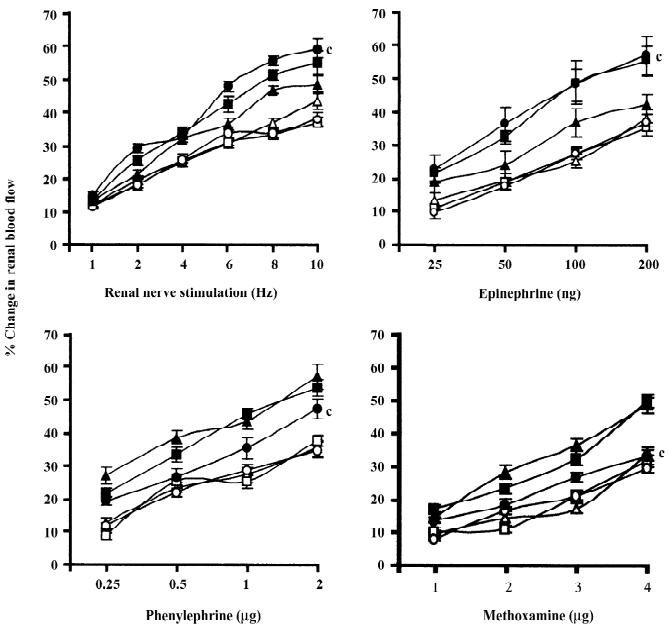
Normal and renal failure Wistar Kyoto rats In the case of renal nerve stimulation, the administration of chloroethylclonidine produced an accentuation (P<0.01) of renal nerve stimulation-induced renal vasoconstrictor responses in the renal failure Wistar Kyoto rats; the magnitudes of these changes were dose-dependent (Figure 2). However, in normal Wistar Kyoto rats, chloroethylclonidine did not cause any marked alterations in such responses. The mixed adrenergic agonist noradrenaline-induced changes were also unaltered by either doses of chloroethylclonidine in these rats (Figure 2). In contrast, there was a marked dose-dependent accentuation (P<0.01) of the noradrenaline-induced renal vasoconstrictor responses in renal failure Wistar Kyoto rats (Figure 2). In renal failure Wistar Kyoto rats, phenylephrine and methoxamine-induced renal vasoconstrictor responses were attenuated by chloroethyl-clonidine (P<0.01). However, these responses in non-renal failure Wistar Kyoto rats were unaltered by chloroethyl-clonidine (Figure 2). The overall mean percentage changes in the renal blood flow of renal failure and non-renal failure Wistar Kyoto rats to different adrenergic stimuli are shown in Table 3.
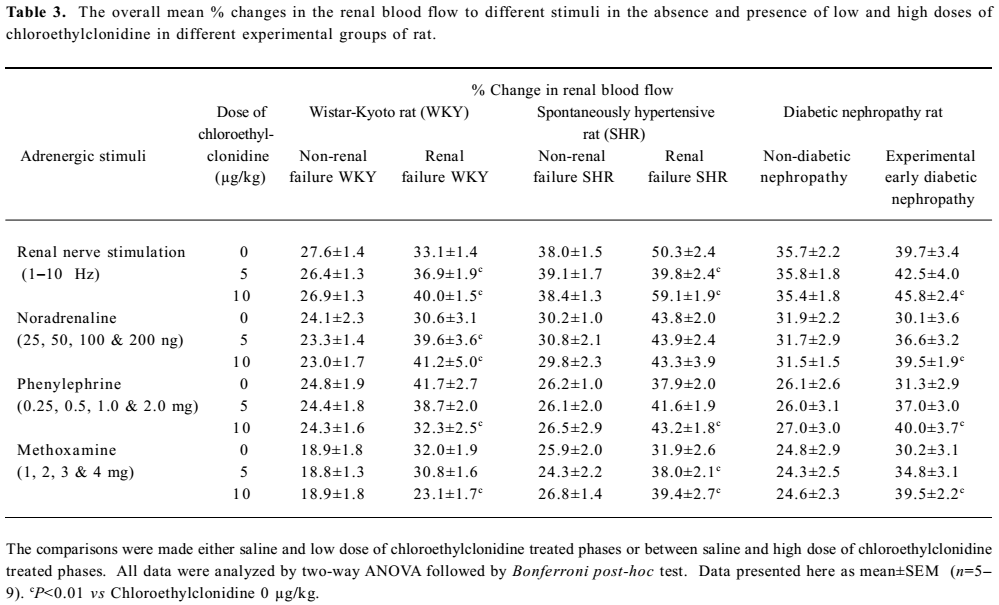
Full table
Spontaneously hypertensive and renal failure spontaneously hypertensive rats Chloroethylclonidine produced interesting results in renal failure spontaneously hypertensive rats with a marked attenuation (by the low dose of chloroethylclonidine) followed by accentuation (by the high dose of chloroethylclonidine) of the renal nerve stimulation-induced renal vasoconstrictor responses (all P<0.01). However, in normal spontaneously hypertensive rats, chloroethylclonidine did not cause any meaningful alteration in the renal nerve stimulation-induced renal vasoconstrictor responses (Figure 3). In the case of noradrenaline-induced vasoconstrictor responses, chloroethyl clonidine at both doses did not canse any alteration (P>0.05) in either of the experimental groups. In the spontaneously hypertensive rats, neither dose of chloroethylclonidine showed any effect on the phenylephrine and methoxamine-induced renal vasoconstrictor responses (P>0.05). Interestingly, in the renal failure spontaneously hypertensive rats, chloroethyl-clonidine in its high dose caused enhancement of phenylephrine and methoxamine-induced renal vasoconstrictor responses (P< 0.01), while its low dose remained insensitive (P>0.05; Figure 3). The overall mean percentage changes in the renal blood flow of renal failure and non-renal failure spontaneously hypertensive rats to different adrenergic stimuli are shown in Table 3.
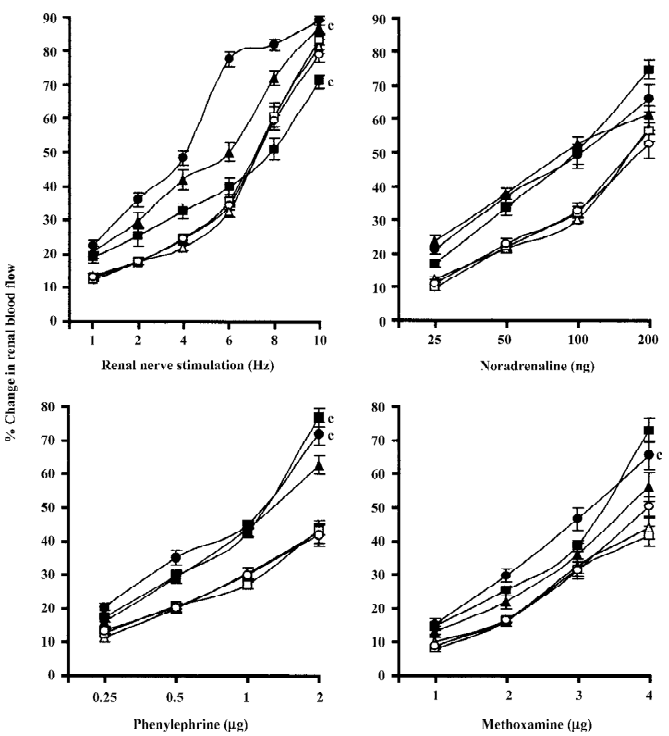
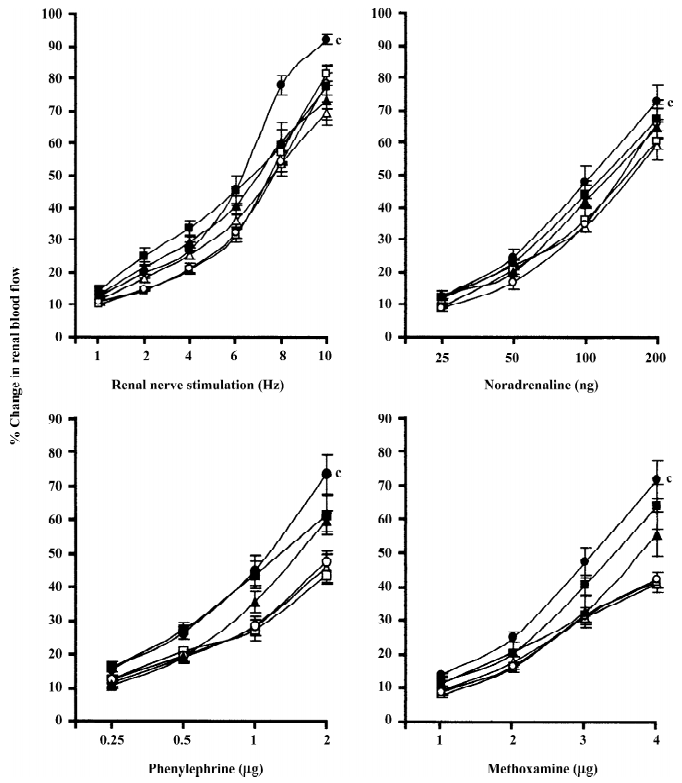
Rats with experimental early diabetic nephropathy In these group of rats chloroethylclonidine accentuated (all P<0.01) all the adrenergically-induced renal vasoconstrictor responses in the rats with experimental early diabetic nephropathy. However, in the non-diabetic nephropathy rats of this particular experimental group chloroethylclonidine did not cause any marked (all P<0.01) shift in these responses (Figure 4). The overall mean percentage changes in the renal blood flow of renal failure and non-diabetic nephropathy and experimental early diabetic nephropathy rats to different adrenergic stimuli are shown in Table 3.
Discussion
This study, perhaps for the first time, shows the functional involvement of chloroethylclonidine sensitive α1B-adrenoceptors in modulating adrenergically-induced renal vasoconstrictions in renal failure, hypertensive renal failure, and early diabetic nephropathy rats. In the rats with normal renal functions, such functional contribution of this α1-adrenoceptor subtype was absent.
In the rats with renal impairment, the adrenergically-induced renal vasoconstrictions were either accentuated or attenuated by chloroethylclonidine, whereas, these responses in the non-renal failure and non-diabetic nephropathy rats remain unchanged. A similar set of observations has been made in several earlier studies on rats with different pathological states, particularly in some types of experimental hypertension, heart failure, diabetes, and also in a combined state of hypertension and diabetes[10,11,14]. Most importantly, these studies have indicated a possible minor functional involvement of α1B-adrenoceptors in the adrenergically-induced renal vasoconstrictions in these rats. In normal rats, however, no such involvement of α1B-adrenoceptors in modulating renal vasoconstrictions has been reported. In this context, the present study investigated whether a similar phenomenon could exist in some important pathological conditions characterized with impaired renal functions and provides further support to the earlier stated view on the interesting functional characteristics of α1B-adrenoceptors in renal vasculature.
The present study showed that in the rats with renal impairment, chloroethylclonidine accentuated the adrenergically-induced renal vasoconstrictor responses, with some exceptions in renal failure Wistar Kyoto rats where chloroethylclonidine attenuated the phenylephrine and methoxamine induced responses. In general, however, there was an enhancement of the renal vasoconstrictor responses elicited by renal nerve stimulation and noradrenaline (renal failure Wistar Kyoto rats); by renal nerve stimulation, phenylephrine, and methoxamine (renal failure spontaneously hypertensive rats), and also by renal nerve stimulation and all the adrenergic agonists (rats with experimental early diabetic nephropathy) in the presence of chloroethylclonidine.
It has been shown that in some vasculature, noradrena-line, which is released due to the stimulation of renal nerves, caused α1A-adrenoceptor subtype-mediated vasoconstriction[16,34]. Like endogenously-released noradrenaline, exogenously administered noradrenaline is also reported to exert its action through α1A-adrenoceptor subtypes with little or no evidence for the involvement of α1B-, α1D-, and even α2-adrenoceptors[16]. As an apparent difference of some of these findings, there are reports on the involvement of α1B- and α1D-adrenoceptors in mediating exogenously administered and endogenously available noradrenaline in in vitro experiments[15,16,35]. However, as asserted in some earlier reported findings, in the presence of chloroethylclonidine and enhanced sympathetic activity, any such involvement of α1B- and α1D-adrenoceptors in mediating renal vasoconstriction will be abolished or impeded. It is stated that chloroethyl-clonidine has selectivity for α1-adrenoceptors in the order of α1B> α1D>> α1A, and that the α1A-adrenoceptor is insensitive to chloroethylclonidine, hence, the α1B- and α1D-adreno-ceptor subtypes will preferentially be inactivated by chloroethylclonidine[36–40] leaving the α1A-adrenoceptors to be acted upon by the adrenergic stimuli, leading to vasoconstrictions. Apart from the possible inactivation by chloroethylclonidine, the suggested inability of the α1D-adrenoceptor subtypes to mediate vasoconstrictions in these pathological states that are characterized with enhanced sympathetic activity, can further be explained based on the observation that the α1D-adrenoceptor subtypes are phosphorylated in the face of enhanced sympathetic activity[4,13,41]. Indeed, the augmented renal vasoconstrictor response is reported in several pathophysiological states (spontaneously hypertensive rats, diabetic hypertensive rats, renal failure rats, and 2K1C Goldblatt hypertensive rats), as reported earlier[10,11,14,17].
With this background, it can be suggested that in the presence of chloroethylclonidine, both renal nerve stimulation and noradrenaline-induced renal vasoconstrictions was mediated by α1A-adrenoceptors in the rats with renal impairment. This view can further be explained based on an earlier report that in these rats, chloroethylclonidine blocked α1B-adrenoceptor subtypes and caused α1A-adrenoceptor subtype-mediated renal vasoconstrictor responses to be accentuated by the endogenous or exogenously administered noradrenaline. It can also be suggested that in these rats there could be an upregulation of α1A-adrenoceptors or involvement of presynaptic α1B-adrenoceptors in determining renal vasoconstriction. In the later case, it can be suggested that when these receptors are blocked by chloroethylclonidine, the presynaptic autoinhibitory feedback is removed and allow more noradrenaline to be released, which consequently resulted in a larger post-synaptic response[22]. Another possible explanation of these observations in the rats with renal impairment could be the downregulation of certain α1-adrenoceptors in the renal vasculature. In support of this view, it is reported that in diabetes and hypertension, the α1B-adrenoceptors could be downregulated by the adrenergic nerves leaving postsynaptic receptors, like the α1A-adrenoceptor (the predominant type in renal vasculature), to be enhanced in order to maintain the effectiveness of the α1-adrenergic nervous system[32,34].
Chloroethylclonidine showed a similar accentuating effect on the phenylephrine and methoxamine-mediated responses in the renal failure spontaneously hypertensive and experimental early diabetic nephropathy rats, while in the non-renal failure and non-diabetic nephropathy rats it was insensitive. These accentuations of renal vasoconstrictions caused by phenylephrine (selective to the α1-adrenoceptor subtypes) and methoxamine (selective to the α1A-and α1D-adrenoceptor subtypes) can be explained in light of our discussion on the accentuation of noradrenaline and renal nerve stimulation-mediated responses in the presence of chloro-ethylclonidine.
In renal failure spontaneously hypertensive rats, it was further observed a biphasic action of chloroethylclonidine on the renal nerve stimulation induced changes. It is reported that there could be a crosstalk relationship between the α1A- and α1B-adrenoceptor subtypes in terms of inhibition of α1A-adrenoceptor activities by α1B-adrenoceptor subtypes[43]. This phenomenon could help us to explain the observed biphasic action of chloroethylclonidine in renal nerve stimulation-induced renal vasoconstrictor responses in renal failure spontaneously hypertensive rats.
In the case of low dose of chloroethylclonidine, the puzzling attenuation of the renal nerve-induced vasoconstrictions in renal failure spontaneously hypertensive rats could be attributed to the blockade of the unsaturated presynaptic α1B-adrenoceptors, and it is widely reported that chloro-ethylclonidine can act presynaptically. In explaining the accentuation the vasoconstrictor responses in these rats by a higher dose of chloroethylclonidine, it is possible that the occupation of the α1B-adrenoceptors leads to an alteration of the properties of α1A-adrenoceptors so that the normal agonist and antagonist interaction can not occur. In this situation, it could be a possibility that the blockade of α1B-adrenoceptors by chloroethylclonidine would enhance the sensitivity of the remaining α1-adrenoceptors (α1A-and α1D-adrenoceptors) so that a potentiation of renal vasoconstrictor responses occurs. Another possibility is that, in this particular pathological state with renal failure and hypertension, there might be an activation of spare receptors due to the blockade of the α1B-adrenoceptors[10,14,17,44].
Together, these data lead us to suggest that in renal failure Wistar Kyoto and spontaneously hypertensive rats, and also in the rats with experimental early diabetic nephropathy, there was a functional involvement of the α1B-adrenoceptors in modulating adrenergically-induced renal vasoconstric-tions. The results obtained also lead us to suggest that there might be a complex interaction between the chloroethylclonidine sensitive α1-adrenoceptor subtypes. In non-renal failure and non-diabetic nephropathy rats, the functional involvement of the α1B-adrenoceptor subtypes was absent.
References
- DiBona GF, Sawin LL. Role of renal α2-adrenergic receptors in spontaneously hypertensive rats. Hypertension 1987;9:41-81.
- Sattar MA, Johns EJ. Evidence for an alpha 1-adrenoceptor subtypes mediating adrenergic vasoconstriction in Wistar normotensive and stroke prone hypertensive rats. J Cardiovasc Pharmacol 1994;23:232-9.
- Yang M, Reese J, Cotecchia S, Michel MC. Murine alpha1-adrenoceptor subtypes. I. Radioligand binding studies. J Pharmacol Exp Ther 1998;286:841-7.
- García-Sáinz JA, Vazuez-Prado J, Medina LC. α1-adrenoceptors: functions and phosphorylation. Eur J Pharmacol 2000;389:1-12.
- Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev 2001;53:319-56.
- Williams TJ, Clarke DE. Characterization of α1-adrenoceptors mediating vasoconstriction to noradrenaline and nerve stimulation in the isolated perfused mesentery of rat. Br J Pharmacol 1995;114:531-6.
- Salomonsson M, Brannstrom K, Arendshorst WJ. α1-adrenoceptor subtypes in rat renal resistance vessels: in vivo and in vitro studies. Am J Physiol 2000;278:F138-47.
- Hirasawa A, Sugawara T, Awaji T, Tsumaya K, Ito H, Tsujimoto G. Subtype-specific differences in subcellular localization of alpha1-adrenoceptors: chlorethylclonidine preferentially alkylates the accessible cell surface alpha1-adrenoceptors irrespective of the subtype. Mol Pharmacol 1997;52:764-70.
- Walsh MP. Regulation of vascular smooth muscle tone. Can J Physiol Pharmacol 1994;72:919-36.
- Abdul Sattar M, Johns EJ. Alpha 1-adrenoceptor subtypes mediating adrenergic vasoconstriction in kidney, one-clip Goldblatt and deoxycorticosterone acetate-salt hypertensive rats. J Cardiovasc Pharmacol 1994;24:420-8.
- Yatsu T, Aoki M, Inagaki O. Preventive effect of zelandopam, a dopamine D1 receptor agonist, on cisplatin-induced acute renal failure in rats. Eur J Pharmacol 2003;461:191-5.
- Villalobos-Molina R, López-Guerrero JJ, Ibarra M. Alpha1D- and alpha1A-adrenoceptors mediate contraction of rat renal artery. Eur J Pharmacol 1997;322:225-7.
- Arévalo-León LE, Gallardo-Ortíz IA, Urquiza-Marín H, Villalobos-Molina R. Evidence for the role of alpha1D- and α1A-adrenoceptors in contraction of rat masentric artery. Vascular Pharmacol 2003;40:91-6.
- Armenia A, Munavvar AS, Abdullah NA, Helmi A, Johns EJ. The contribution of adrenoceptor subtypes in the renal vasculature of diabetic spontaneously hypertensive rats. Br J Pharmacol 2004;142:719-26.
- Jahnichen S, Eltze M, Pertz HH. Evidence that alpha (1B)-adrenoceptors are involved in noradrenaline-induced contractions of rat tail artery. Eur J Pharmacol 2004;488:157-67.
- Zacharia J, Hillar C, MacDonald A. α1-adrenoceptor subtypes involved in vasoconstrictor responses to exogenous and neurally released in rat femoral resistance arteries. Br J Pharmacol 2004;141:915-24.
- Khan AH, Sattar MA, Abdullah NA, Johns EJ. Impact of renal failure on the α1-adrenoceptor subtype mediating vasoconstriction in the kidney of the rat. Eur J Pharmacol 2007;569:110-8.
- Abbas SA, Munavvar AS, Abdullah NA, Johns EJ. Involvement of α1-adrenoceptor subtypes in the cardiac failure in spontaneously hypertensive rats. J Basic Applied Sci 2006;2:59-69.
- Stassen FR, Willemsen MJ, Janssen GM, DeMey JG. Alpha 1-adrenoceptor subtypes in rat aorta and mesenteric small arteries are preserved during left ventricular dysfunction post-myocardial infarction. Cardiovasc Res 1997;33:706-13.
- Kang DG, Lee AS, Mun YJ, Woo WH, Kim YC, Sohn EJ, et al. Butein ameliorates renal concentrating ability in cisplatin-induced acute renal failure in rats. Biol Pharm Bull 2004;27:366-70.
- Yatsu T, Aoki M, Inagaki O. Preventive effect of zelandopam, a dopamine D1 receptor agonist, on cisplatin-induced acute renal failure in rats. Eur J Pharmacol 2003;461:191-5.
- Shimeda Y, Hirotani Y, Akimoto Y, Shindou K, Ijiri Y, Nishihori T, et al. Protective effects of capsaicin against cisplatin-induced nephrotoxicity in rats. Biol Pharm Bull 2005;28:1635-8.
- Saad SY, Al-Rikabi AC. Protection effects of taurine supplementation against cisplatin-induced nephrotoxicity in rats. Chemotherapy 2002;48:42-8.
- Yin XX, Zhang YD, Shen JP, Wu HW, Zhu X, Li LM, et al. Protective effects of bendazac lysine on early experimental diabetic nephropathy in rats. Acta Pharmacol Sin 2005;26:721-8.
- Saad SY, Arafah MM, Najjar TA. Effects of mycophenolate mofetil on cisplatin-induced renal dysfunction in rats. Cancer Chemother Pharmacol 2007;59:455-60.
- Cooper ME, Allen TJ, Jerums G, Doyle AE. Accelerated progression of diabetic nephropathy in the spontaneously hypertensive streptozo-tocin diabetic rat. Clin Exp Pharmacol Physiol 1986;13:655-62.
- Usui H, Shikata K, Matsuda M, Okada S, Ogawa D, Yamashita T, et al. HMG-CoA reductase inhibitor ameliorates diabetic nephropathy by its pleiotropic effects in rats. Nephrol Dial Transplant 2003;18:265-72.
- Yin X, Zhang Y, Wu H, Zhu X, Zheng X, Jiang S, et al. Protective effects of Astragalus saponin I on early stage of diabetic nephropathyin rats. J Pharmacol Sci 2004;95:256-66.
- Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001;44:1957-72.
- Nakano R, Kurosaki E, Shimaya A, Kajikawa S, Shibasaki M. YM440, a novel hypoglycemic agent, protects against nephropathy in Zucker fatty rats via plasma triglyceride reduction. Eur J Pharmacol 2006;549:185-91.
- Cruzado JM, Lloberas N, Torras J, Riera M, Fillat C, Herrero-Fresneda I, et al. Regression of advanced diabetic nephropathy by hepatocyte growth factor gene therapy in rats. Diabetes 2004;53:1119-27.
- Ibarra M, Pardo JP, López-Guerrero JJ, Villalobos-Molina R. Differential response to chloroethylclonidine in blood vessels of normotensive and spontaneously hypertensive rats: role of alpha 1D- and alpha 1A-adrenoceptors in contraction. Br J Pharmacol 2000;129:653-60.
- Perez DM, Piascik MT, Malik N, Gaivin R, Graham RM. Cloning, expression, and tissue distribution of the rat homolog of the bovine alpha 1C-adrenergic receptor provide evidence for its classification as the alpha 1A subtype. Mol Pharmacol 1994;46:823-31.
- Jahnichen S, Eltze M, Pertz HH. Evidence that alpha (1B)-adrenoceptors are involved in noradrenaline-induced contractions of rat tail artery. Eur J Pharmacol 2004;488:157-67.
- Williams TJ, Clarke DE. Characterization of α1-adrenoceptors mediating vasoconstriction to noradrenaline and nerve stimulation in the isolated perfused mesentery of rat. Br J Pharmacol 1995;114:531-6.
- Leclerc G, Rouot B, Schwartz J, Velly J, Wermuth CG. Studies on some para-substituted clonidine derivatives that exhibit an alpha-adrenoceptor stimulant activity. Br J Pharmacol 1980;71:5-9.
- Docherty JR, O’Rourke M. The α-adrenoceptor-mediated actions of chloroethylclonidine. Gen Pharmacol 1997;28:97-101.
- Hirasawa A, Sugawara T, Awaji T, Tsumaya K, Ito H, Tsujimoto G. Subtype-specific differences in subcellular localization of alpha1-adrenoceptors: chlorethylclonidine preferentially alkylates the accessible cell surface alpha1-adrenoceptors irrespective of the subtype. Mol Pharmacol 1997;52:764-70.
- Yang M, Reese J, Cotecchia S, Michel MC. Murine alpha1-adrenoceptor subtypes. I. Radioligand binding studies. J Pharmacol Exp Ther 1998;286:841-7.
- Ibarra M, Lopez-Guerrero JJ, Villalobos-Molina R. The influence of chloroethylclonidine-induced contraction in isolated arteries of Wistar Kyoto rats: alpha1D- and alpha1A-adrenoceptors, protein kinase C, and calcium influx. Arch Med Res 2001;32:258-62.
- García-Sáinz JA, Vazuez-Prado J, Medina LC. α1-adrenoceptors: functions and phosphorylation. Eur J Pharmacol 2000;389:1-12.
- Li Z, Silver WP, Koman LA, Strandhoy JW, Rosencrance E, Gordon S, et al. Role of alpha-1 adrenoceptor subtypes mediating constriction of the rabbit ear thermoregulatory microvasculature. J Orthop Res 2000;18:156-63.
- Kahan T. Prejunctional adrenergic receptors and sympathetic neurotransmission: studies in canine skeletal muscle vasculature in situ. Acta Physiol Scand 1987;560:1-38.
- Piascik MT, Spark S MS, Pruitt TA, Soltis EE. Evidence for a complex interaction between the subtypes of the α1-adrenoceptor. Eur J Pharmacol 1991;199:279-89.