
Phosphorylation of β-actin by protein kinase C-delta in camptothecin analog-induced leukemic cell apoptosis1
Introduction
Apoptosis or programmed cell death is generally characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms, which is an essential physiological process throughout the life of multi-cellular organisms[1]. Apoptosis is involved in development, elimination of damaged cells, and maintenance of tissue homeostasis as well as pathogenesis and therapeutics of diseases[2]. For example, one of the most important advances in cancer research in recent years is the recognition that apoptosis is crucially involved in the regulation of tumor formation and treatment response[3]. Apoptosis induction is frequently used in clinical oncology, such as chemotherapy, gamma-irradiation and immunotherapy. Vice versa, failure to undergo apoptosis may result in treatment resistance[4]. Thus, understanding how apoptosis is regulated in response to anticancer chemotherapy and how cancer cells evade apoptotic death provides novel opportunities for a more rational approach to develop molecular-targeted therapies for cancers[3–5].
Camptothecin is an alkaloid isolated from the Chinese tree Camptotheca acuminate, which targets the intranuclear enzyme topoisomerase I and advances to the forefront of several areas in clinical and preclinical trials of therapeutic chemotherapy[6,7]. NSC606985, a new water-soluble camptothecin ester derivative, was reported to induce acute myeloid leukemia (AML) cells to undergo apoptotic cell death at nanomolar concentrations by mediating proteolytic activation of protein kinase Cδ (PKCδ) [8]. Furthermore, potential therapeutic effects of NSC606985 was also shown in a leukemic mice model [9]. Using two-dimensional electrophoresis (2-DE) combined with MALDI-TOF/TOF tandem mass spectrometry to analyze sub-cellular protein expression profiles of NSC606985-induced apoptotic AML cells, a set of deregulated proteins were recently identified [10]. To find more proteins involved in NSC606985-induced apoptosis, this study performed a differential proteomic analysis with the total proteins of U937 cells treated with or without NSC606985. We found that β-actin was phosphorylated following PKCδ activation in leukemic U937 cells, which would shed new insights on the molecular mechanisms of NSC606985-induced apoptosis.
Materials and methods
Cell line and treatment AML U937 cells were cultured in RPMI-1640 medium (Sigma, St Louis, MI) supplemented with 10% fetal calf serum (Gibco BRL, Gaithersburg, MD) in a 5% CO2/95% air humidified atmosphere at 37oC. For experiments, U937 cells were seeded at 3×105 cells/ml and incubated with 50 nmol/L NSC606985 (kindly provided by National Cancer Institute Anticancer Drug Screen standard agent database, Bethesda, MD) with or without the PKCδ-specific inhibitor of 1 µmol/L rottlerin (BIOMOL, Plymouth, PA). Cell viability was determined by the trypan-blue exclusion assay, and growth inhibition rate was calculated according to viable cell numbers of treated cells against numbers of untreated cells. For morphologic observation, cells were collected onto slides by cytospin (Shandon, Runcorn, United Kingdom), stained with Wright staining, and examined under light microscope. To assess apoptosis, annexin-V assay was performed by the ApoAlert Annexin V kit (BD Biosciences, Palo Alto, CA, USA) on flow cytometry (BD FACSCalibur, Palo Alto, CA, USA).
Protein preparation U937 cells (about 1×107) were harvested and washed for 3 times with Tris-buffered sucrose (0.25 mol/L sucrose, 10 mmol/L Tris-HCl, pH 7.4). Then, cell pellets were dissolved in lysis buffer containing 7 mol/L urea, 2 mol/L thiourea, 4% m/v CHAPS, 50 mmol/L DTT, 40 mmol/L Tris-Base, 0.2% Bio-lyte (pH 3-10), 10% isopropanol, 12.5% water-saturated isobutanol, and 1% protease inhibitor cocktail for 1 h at room temperature, followed by centrifugation (35 000×g, 1 h) at 4 ℃. The supernatant was quantified using Bio-Rad RC DC protein assay kit (Bio-Rad, Hercules, CA) and aliquoted. The protein samples were stored at -80 ℃ until analysis.
For the dephosphorylation of phosphoproteins with alkaline phosphatase (AP), 200 μg cardiac protein and 20 U calf intestine AP (Promega, Madison, WI) were incubated in 300 µL of AP reaction buffer (50 mmol/L Tris HCl pH 8.2, 150 mmol/L NaCl, 1 mmol/L MgCl2, 1% v/v protease inhibitor cocktail) at 30 ℃ for 60 min. Control samples were incubated in the same buffer with no enzyme. After incubation the proteins were applied to 2-DE followed by Western blot.
Western blot Proteins 30 µg were loaded onto 10% SDS-PAGE for separation, and then electrophoretically transferred to NC membrane (Bio-Rad, Hercules, CA). The blots were stained with 0.2% Ponceau S red to ensure equal protein loading. After blocking with 5% nonfat milk in TBS, the membranes were probed with anti-PKCδ (1:3000, Santa Cruz, CA), cleaved caspase-3 (1:500; Dako Cytomation, Denmark), poly(ADP [adenosine diphosphate]–ribose) polymerase (PARP; 1:500, Santa Cruz, CA), and β-actin (1:10000, Oncogene, San Diego, CA), followed by horseradish peroxidase (HRP)–linked secondary antibodies (Dako Cytomation, Denmark). The signals were detected by chemiluminescence phototope-HRP kit (Cell Signaling) according to manufacturer’s instructions.
2-DE and image analysis[11] After diluted in 300 µL rehydration buffer containing (8 mol/L urea, 2% m/v CHAPS, 25 mmol/L DTT, 0.2% Bio-lyte (3-10, pI range) and 0.002% bromophenol blue), 160 µg proteins were applied onto 17 cm IPG strips (NL, pH 3-10, Bio-Rad, Hercules, CA, USA). The first dimension was carried out on a Protean IEF Cell system (Bio-Rad, Hercules, CA) at 20 ℃ with following schedules: 50 V for 12 h, 200 V for 30 min, 500 V for 1 h, 1 000 V for 1 h, 10 000 V for 2 h and followed by 10000V for 60 000V h. After IEF, the IPG strips were equilibrated in a buffer (6 mol/L urea, 20% glycerol, and 2% SDS in 0.05 mol/L Tris-HCl buffer, pH 8.8) containing with 1% w/v DTT (step I) and 4% w/v iodoacetamide (IAA) (step II) iodoacetamide respectively. The second dimensional separation was carried out on 12.5% SDS-PAGE, followed by the silver staining. Silver stained gels were scanned using GS-800 calibrated imaging density meter (Bio-Rad, Hercules, CA, USA). The spots were analyzed using PDQuest Image Analysis Software version 7.2 (Bio-Rad, Hercules, CA, USA).
In-gel digestion and mass spectrometry The protein spots were cut out of 2-D gels using Gelpix Spot-Excision Robot (Genetix, Hampshire, UK), digested and identified as previously described[12]. Shortly, those protein spots destained with equal volume of 30 mmol/L potassium ferricyanide and 100 mmol/L sodium thiosulfate, followed by washing with Milli-Q water, 25 mmol/L ammonium bicarbonate/50% ACN and 100% ACN. Then the gel pieces were dried and digested overnight in 10 µL of trypsin (4 ng/µL, Trypsin Gold, Promega, Madison, WI) in 25 mmol/L ammonium bicarbonate at 37 ℃. The extracted tryptic peptides were lyophilized and resuspended in 1 μL matrix solution containing 5 mg/mL α-cyano-4- hydroxycinnamin acid prepared in 50% ACN/0.1% TFA, followed by spotting onto the MALDI sample target plate and identification with MALDI-TOF-TOF mass spectrometer (4700 Proteomics Analyzer, Applied Biosystem, Foster City, CA).
Rhodamine-phalloidin staining Cells in glass-bottom dishes were fixed in 40 g/L paraformal-dehyde (pH 7.2) for 10 min at 4 ℃. The cells were rinsed with PBS twice, then permeabilized for 10 min in 2 g/L Triton X-100 (Sigma, St Louis, MI) at room temperature, and nonspecific background was blocked using 5 g/L BSA (Sigma, St Louis, MI) for 1 h. The cells were then stained with 5 µg/mL fluorescein rhodamine-phalloidin (Molecular Probes, Eugene, OR, USA) at room temperature for 40 min. After that, the cells were rinsed with PBS twice and stained with 5 µg/L DAPI (Molecular Probes, Eugene, OR). All the above procedures performed in dark. Digital images of fluorescent slides were viewed under a laser scanning confocal microscope (Carl Zeiss LSM-510, Jena, Germany). Rhodamine-phalloidin mean fluorescence intensity per cell was used as a measure of F-actin concentration.
Statistical analysis The values were expressed as mean±SD. The student t test was used for statistical analysis between two groups. Significant level was set at P<0.05.
Results
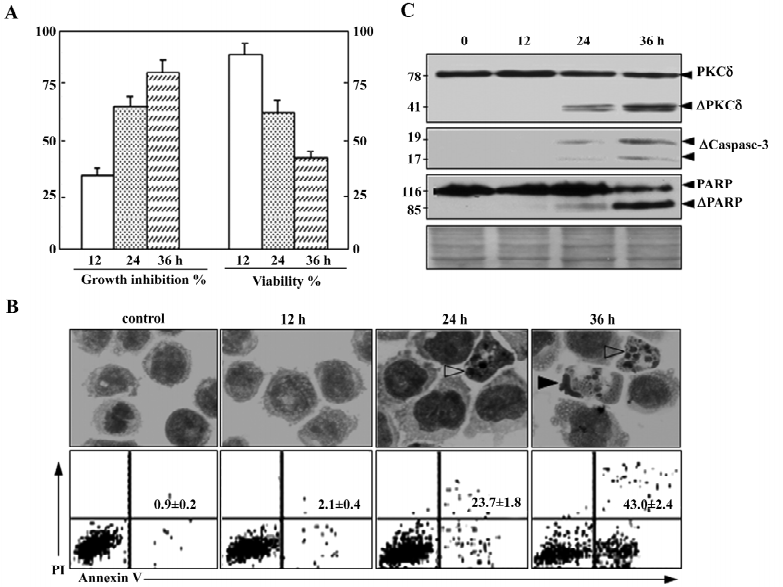
As described previously, 50 nmol/L NSC606985 inhibited growth (Figure 1A, left) and reduced viability of U937 cells(Figure 1A, right). Accordingly, these treated cells underwent apoptosis as evidenced by morphologic changes (top panels, Figure 1B), annexin-V assay (bottom panels, Figure 1B) as well as activation of caspase-3 and cleavage of its substrate PARP (Figure 1C). For protein profile analysis, protein extracts of U937 cells treated with and without 50 nmol/L of NSC606985 for 36 h were applied to 2-DE, followed by analysis of PDQuest software. The overall protein expression profiles between pI 3–10 and MW 10–80 kDa had a high resolution and reproducibility. A pair of representative 2-DE gel images was shown (Figure 2). Protein spots with significant changes in densities (Student t-test, P<0.05) in a consistent direction (increase or decrease) were judged as deregulated ones, which were cut out for identification.
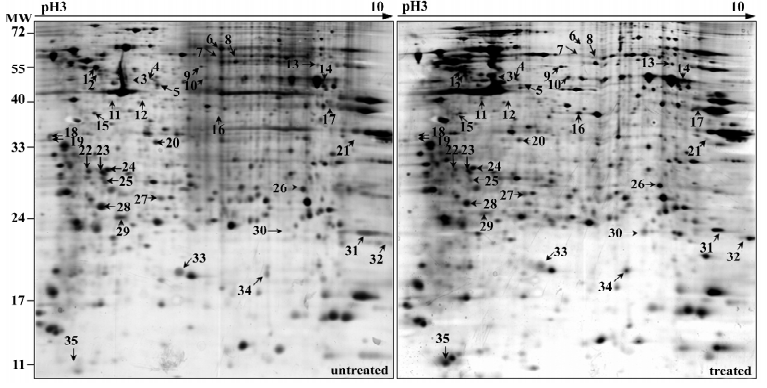
Totally, 35 up- or down-regulated spots were successfully identified by MALDI-TOF/TOF mass spectrometry with PMF and/or MS/MS followed by database searching, as shown in Figure 2 and Table 1. They included 28 unique proteins and these proteins were classified into cytoskeleton, RNA metabolism, DNA metabolism, protein metabolism, carbohydrate metabolism, energy metabolism, signal transduction, stress response, oxidation and reduction, differentiation and others according to their primary functions.
Full table
Notably, cytoskeleton proteins occupied a proportion of these differential proteins (Table 1). As an example, β-actin, a key cytoskeleton protein usually as internal loading control in the Western blot analysis, was demonstrated to be significantly changed on the differential 2-DE analysis. After NSC606985 treatment, β-actin was found to be up-regulated at spot 11 and down-regulated at spot 12 on the 2-DE map (Figure 2 and 3A), while the total β-actin protein was equal between the untreated and treated cells (Figure 3B). Furthermore, immunoblot analysis of β-actin of untreated U937 cells on the 2-DE map also showed that β-actin had two spots, which share similar molecular weight and different pI, and the spot at the basic-end disappeared while the one at acid-end increased after NSC606985 treatment (Figure 3C, top two panels), which was consistent with the proteomic analysis (Figure 3A).
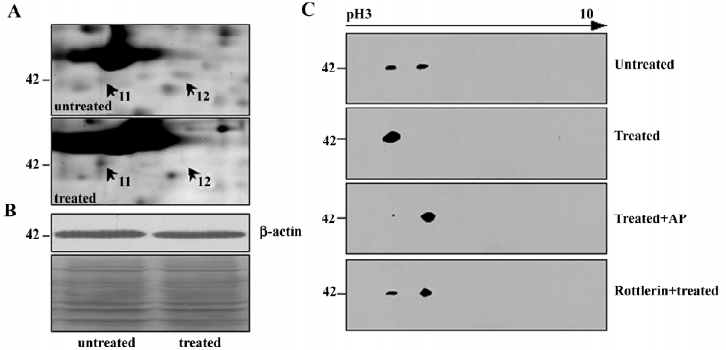
It is known that phosphorylation is one of the most important reversible post-translational modifications (PTMs) and it would cause protein acidification and shitting to the acid-end on the 2-DE map[13]. Thus, we presumed that β-actin had been phosphorylated during apoptosis induction. To ascertain this, the protein extract of NSC606985-treated cells was incubated with alkaline phosphatase (AP) which can cause dephosphorylation from the proteins. As shown in the third panel of Figure 3C, most of β-actin transferred to the basic end after dephosphorylation by the AP treatment, supporting that β-actin was phosphorylated during the NSC606985-induced apoptotic process.
Previously, it was reported that cleaved PKCδ plays a key role in NSC606985-induced apoptotic process[8]. We extrapolated whether phosphorylation of β–actin is mediated by activated PKCδ protein. Therefore, rottlerin, a specific inhibitor of PKCδ, was used to pretreat U937 cells. The results showed that rottlerin could inhibit the phosphorylation of β–actin in the apoptotic process (Figure 3C, the bottom panel), indicating that the phosphorylation of β-actin induced by NSC606985 treatment was a downstream event of PKCδ activation.
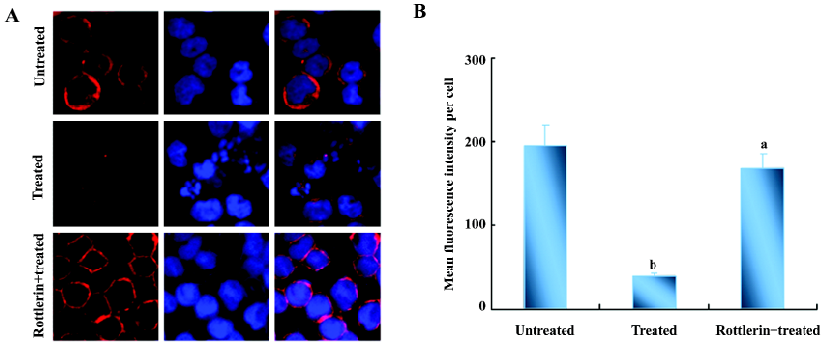
As reported[14], phosphorylated actin monomers (globular actin, G-actin) did not polymerize even under conditions favorable for actin polymerization. Thus, the amount of polymerized actin (fibrous actin, F-actin) would decrease when actin is phosphorylated. We speculated that the amount of F-actin in U937 cells was reduced after NSC606985 treatment. Then we detect the change of actin organization with hodamine-phalloidin, which can specifically bind and stabilize F-actin[15]. The fluorimetric studies indicated that the content of F-actin per cell was reduced significantly after NSC66985 treatment (top two panels of Figure 4A and Figure 4B), which could be rescued by rottlerin pretreatment (Figure 4A, the bottom panel and Figure 4B).
Discussion
It was known that the abnormality of cytoskeleton protein is one of the fundamental changes during apoptosis[16]. Alteration of the cytoskeleton frequently results in membrane blebbing in apoptotic cells[17,18]. Meanwhile, members of cytoskeleton proteins are frequently used as drug target. For example, microtubule is an important target for anti-cancer chemotherapeutic agents for the treatment of cancer, such as vinca alkaloids and taxanes[17]. Here sets of proteins were found to be altered after NSC606985 treatment and cytoskeleton proteins were among the most significant alteration in the treatment, which possibly indicated that cytoskeleton modulation were involved in the action of NSC606985. As a support, Chang et al[19] reported that BPR0Y007, a novel topoisomerase I inhibitor, could have dual inhibition of topoisomerase I and tubulin polymerization. However, it is still under investigation that the alteration of skeleton proteins induced by NSC606985 treatment is a trigger mechanism or just a late event in the apoptotic process.
Actin filaments provide the basic infrastructure for maintaining cell morphology and functions such as adhesion, motility, exocytosis, endocytosis, and cell division[20]. Notably, after the NSC606985 treatment, β-actin was found to be phosphorylated, which was reported to regulate the polymerization of β-actin[14,20]. For example, by viscometry and electron microscopy, Seij et al[14] had demonstrated that G-actin could not polymerize to F-actin (polymerized actin) when amoeba G-actin was phosphorylated. Meanwhile, F-actin disorganization was thought to be closely associated cell apoptosis in that cells with a fragmented nucleus displayed completely disorganized F-actin[18] . Intriguingly, the amount of F-actin in U937 cells was found to decrease after NSC606985 treatment in this study. We speculated that the β-actin was phosphorylated and then F-actin was apt to disorganized by the NSC606985 treatment.
PKCδ, a ubiquitously expressed member of the novel PKC family, is activated by translocation, tyrosine phosphorylation, or proteolytic cleavage into 41-kDa catalytically active fragment[21] . The isoenzyme enigmatically presents the multifunctional properties and is implicated in the regulation of a variety of cellular processes, including secretion, cell cycle progression, apoptosis, differentiation, and tumor development[22] . As previously reported, PKCδ activation was an upstream and critical event in NSC606985-induced apoptosis. In the present study, we found that β-actin was phosphorylated in the experimental group and the event was downstream of PKCδ activation. In accordance, Carole et al[23] found co-localization of PKCδ with nonmuscle actin in airway epithelial cells and binding of PKCδ to actin was concentration dependent and enhanced by the presence of PKC activators. In addition, β-actin had PKCδ recognizing motif (forecasting by scansite software, date not shown). It will be meaningful to investigation that they were phosphorylated by PKCδ directly and/or indirectly.
In summary, based on our previous report that nanomolar concentration of camptothecin analog NSC606985 induces U937 cells to undergo apoptotic cell death, this work used 2-DE combined with MALDI-TOF/TOF to perform proteomic analysis of apoptotic U937 cells. As a result, we found a series of deregulated cytoskeleton proteins. Moreover, β-actin was found to be phosphorylated and it was downstream events of PKCδ activation, which will shed new insights for understanding the mechanisms of the camptothecin-induced apoptosis.
References
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35:495-516.
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 1999;59:1391-9.
- Vermeulen K, Van Bockstaele DR, Berneman ZN. Apoptosis: mechanisms and relevance in cancer. Ann Hematol 2005;84:627-39.
- Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004;23:2934-49.
- Chen GQ, Wang LS, Wu YL, Yu Y. Leukemia, an effective model for chemical biology and target therapy. Acta Pharmacol Sin 2007;28:1316-24.
- Li QY, Zu YG, Shi RZ, Yao LP. Review camptothecin: current perspectives. Curr Med Chem 2006;13:2021-39.
- Bomgaars L, Berg SL, Blaney SM. The development of camptothecin analogs in childhood cancers. Oncologist 2001;6:506-16.
- Song MG, Gao SM, Du KM, Xu M, Yu Y, Zhou YH, et al. Nanomolar concentration of NSC606985, a camptothecin analog, induces leukemic-cell apoptosis through protein kinase Cdelta-dependent mechanisms. Blood 2005;105:3714-21.
- Liu W, Zhu YS, Guo M, Yu Y, Chen GQ. Therapeutic efficacy of NSC606985, a novel camptothecin analog, in a mouse model of acute promyelocytic leukemia. Leuk Res 2007;31:1565-74.
- Yu Y, Wang LS, Shen SM, Xia L, Zhang L, Zhu YS, et al. Subcellular proteome analysis of camptothecin analogue NSC606985-treated acute myeloid leukemic cells. J Proteome Res 2007;6:3808-18.
- Han YH, Xia L, Song LP, Zheng Y, Chen WL, Zhang L, et al. Comparative proteomic analysis of hypoxia-treated and untreated human leukemic U937 cells. Proteomics 2006;6:3262-74.
- Jin X, Xia L, Wang LS, Shi JZ, Zheng Y, Chen WL, et al. Differential protein expression in hypertrophic heart with and without hypertension in spontaneously hypertensive rats. Proteomics 2006;6:1948-56.
- Mukherji M. Phosphoproteomics in analyzing signaling pathways. Expert Rev Proteomics 2005;2:117-28.
- Sonobe S, Takahashi S, Hatano S, Kuroda K. Phosphorylation of Amoeba G-actin and its effect on actin polymerization. J Biol Chem 1986;261:14837-43.
- Kwiatkowska K, Sobota A. Engagement of spectrin and actin in capping of FcgammaRII revealed by studies on permeabilized U937 cells. Biochem Biophys Res Commun 1999;259:287-93.
- Janmey PA. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiol Rev 1998;78:763-81.
- Attard G, Greystoke A, Kaye S, De Bono J. Update on tubulin-binding agents. Pathol Biol 2006;54:72-84.
- Van de Water B, Kruidering M, Nagelkerke JF. F-actin disorganization in apoptotic cell death of cultured rat renal proximal tubular cells. Am J Physiol 1996;270:F593-603.
- Chang JY, Hsieh HP, Pan WY, Liou JP, Bey SJ, Chen LT, et al. Dual inhibition of topoisomerase I and tubulin polymerization by BPR0Y007, a novel cytotoxic agent. Biochem Pharmacol 2003;65:2009-19.
- Rao J, Li N. Microfilament actin remodeling as a potential target for cancer drug development. Curr Cancer Drug Targets 2004;4:345-54.
- Jackson DN, Foster DA. The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J 2004;18:627-36.
- Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med 2003;7:341-50.
- Liedtke CM, Hubbard M, Wang X. Stability of actin cytoskeleton and PKC-delta binding to actin regulate NKCC1 function in airway epithelial cells. Am J Physiol Cell Physiol 2003;284:C487-96.