
Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells
Introduction
Autophagy is a major cellular pathway for the degradation of long-lived proteins and organelles in eukaryotic cells[1]. A large number of intracellular/extracellular stimuli, including amino acid starvation and invasion of microorganisms, are able to induce the autophagic response in cells. In addition to the degradation of cellular proteins and organelles, autophagy can serve as a temporary survival strategy to defend against damage caused by environmental changes through the sequestration of noxious substance (such as certain aggregation-prone proteins or the expression of aggregating mutant variants of specific proteins) and injured organelles. Furthermore, autophagy may also be a strategy for self-destruction through the induction of programmed cell death (PCD), which is different from apoptosis[2].
PCD is an essential and highly orchestrated process, which plays an important role in the development, cellular homeostasis, and prevention of cancer cell growth. There are five types of PCD that have been described: apoptosis, autophagy, anoikis, amorphosis, and mitotic catastrophe[3]. The two main PCD types are referred to as apoptosis (PCD I) and autophagy (PCD II). Apoptosis, or PCD I, is characterized by cell shrinkage, oligonucleosomal DNA fragmentation, chromatin condensation, pyknotic nuclei, and controlled disintegration of the cell into so-called apoptotic bodies. By contrast, autophagy, or PCD type II, is featured by the accumulation of autophagic vacuoles (AV) in the cytoplasm[4].
Many reports have shown that under certain conditions, autophagy and apoptosis appear to interact with each other either positively or negatively[5] in the following ways: (i) autophagy may be indispensable for apoptosis occurrence. Autophagy inhibitors, such as 3-methyladenine (3-MA), delay apoptosis while converse, broad-range caspase inhibitors fail to inhibit autophagy[6,7]; (ii) autophagy may antagonize apoptosis. The inhibition of autophagy may increase the sensitivity of cells to apoptotic signals[8]; and (iii) apoptosis and autophagy may occur independent of each other. The inhibition of apoptosis may convert cell death morphology to autophagic and vice versa[5,9–11].
Many studies have shown that the compromise of autophagic activity may contribute to the development of cancers. In cancer cell lines and experimental carcinogenesis in animals, the autophagy activity was often lower than that in their normal counterparts. Moreover, autophagy could be reactivated by several anticancer agents[12,13]. As2O3 has been proven to be a useful drug for the treatment of acute promyelocytic leukemia (APL). Apoptosis seems to be a main mechanism underlying its antitumor activity[14,15]. Recently, it was shown that As2O3 induced autophagy, but not apoptosis in human malignant glioma cell lines by the upregulation of mitochondrial cell death protein Bcl-2/adenovirus E1B 19 kDa-interacting protein 3[16,17]. As2O3 has also been demonstrated to activate autophagy and induce the autophagic death of MoIt-4 cells through the upregulation of beclin 1[18]. We have previously found that autophagy plays different roles in the crotoxin-induced death of chronic myeloid leukemia cell line K562 cells and human breast cancer cell line MCF-7 cells[19,20]. In the present study, we investigated the role of autophagy in the As2O3-induced death of human acute myeloid leukaemia cell line HL60 cells and its interactions with the apoptotic cascade. The results showed that blocking autophagy before and after As2O3 exposure had opposite effects on the As2O3-induced death of HL60 cells, suggesting that autophagy plays different roles in the different stages of apoptosis.
Materials and methods
Cell culture HL60 leukemia cells were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were maintained in RPMI-1640 medium (Gibco, Rockville, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (Hangzhou Sijiqing Biological Engineering Materials, Hangzhou, China) and 0.03% L-glutamine (Sigma, St Louis, MO, USA) and incubated in a humidified 5% CO2 incubator at 37 ℃. The cells in the mid-log phase were used in the experiments.
Quantitative analysis of the viability of HL60 cells after exposure to As2O3 As2O3 (0.625, 1.25, 2.5, 5, 10, and 20 µmol/L; Sigma, USA) was added to the culture medium, and HL60 cell viability was determined with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay 12, 24, 48, and 72 h after exposure to As2O3 following a previously described method[21]. Briefly, MTT (Sigma, USA) solution was added to the culture medium (final concentration =500 µg/mL) 4 h before the end of the treatment. The reaction was terminated by the addition of 10% acidified SDS (100 µL) to the cell culture. The absorbance value (A) was measured at 570 nm using a multiwell spectrophotometer (Bio-Rad, Richmond CA, USA). The percentage of cell death was calculated with the following formula: cell death (%)=(1-A of experiment well/A of control well)×100%.
Visualization of monodansylcadaverine-labeled AV AV were detected using a method as previously described[22]. The cells were seeded onto 24-well plates and incubated with As2O3 (10 µmol/L) for 3, 6, and 12 h. The cells were then incubated with monodansylcadaverine (MDC; 50 µmol/L) in RPMI-1640 at 37 ℃ for 10 min. After incubation, the cells were washed three times with phosphate buffered saline (PBS) and immediately analyzed with a fluorescence microscope (Eclipse TE 300; Nikon, Japan) equipped with a filter system (V-2A excitation filter: 380/420 nm, barrier filter: 450 nm)[23]. Images were captured with a charge-coupled device (CCD) camera and imported into Adobe Photoshop (Adobe, Japan).
Transmission electron microscopic analyses of autophago-somes HL60 cells were cultured in 60 mm dishes and treated with As2O3 (10 µmol/L) for 6, 12, and 24 h. After treatment, the cells were fixed in ice-cold 2.5% glutaraldehyde in 0.1 mol/L PBS and preserved at 4 ℃ for further processing. When processing resumed, the cells were post-fixed in 1% osmium tetroxide in the same buffer, dehydrated in graded alcohols, embedded in Epon 812, sectioned with ultramicrotome, and stained with uranyl acetate and lead citrate. The sections were examined with a transmission electron microscope (TEM; Technai 10; Philips, the Netherlands).
Cytotoxicity assay It has been reported that lactose dehydrogenase (LDH) leakage not only occurs during necrosis, but also during the process of apoptosis[24,25]. Since 3-MA interferes with the MTT assay, LDH leakage was assessed as an index of cell death after the cotreatment of As2O3 and 3-MA or NH4Cl. To examine the contribution of autophagy to the As2O3-induced death of HL60 cells, the cells were treated with the specific autophagy inhibitor 3-MA (Sigma, USA; 10 mmol/L) or lysosome-neutralizing agent NH4Cl (10 mmol/L) 1 h before As2O3 and LDH leakage was measured 24 h after As2O3, or the cells were treated with 3-MA (10 mmol/L) 5 min to 12 h after As2O3 (10 µmol/L) and LDH leakage was measured 24 h after As2O3. Briefly, after drug treatment, the supernatant of the cell culture was reserved. The cells were rinsed with PBS and lysed with 1% Triton X-100 at 37 ℃ for 45 min. Then samples of supernatants and cell lysates were prepared following the manufacturer’s instructions for the LDH test kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The absorbance value (A) at 400 nm was determined with an automatic multiwell spectrophotometer (Bio-Rad, USA). LDH leakage was calculated as follows: LDH leakage (%)=(A positive/A positive blank)/(A negative/A negative blank)×100%[26].
Immunofluorescence detection of microtubule-associated protein 1 light chain 3 Briefly, the HL60 cells were incubated in 6-well plates and the specific autophagy inhibitor 3-MA (10 mmol/L) or lysosome-neutralizing agent NH4Cl (10 mmol/L) was added 1 h before As2O3 (10 µmol/L); or cells were treated with 3-MA (10 mmol/L) 30 min after As2O3 (10 µmol/L). After being incubated with As2O3 for 24 h, the cells were washed with PBS and then fixed with paraformaldehyde (4% w:v). After rinsing in PBS, the cells were blocked with 0.1% Triton X-100 and 1% bovine serum albumin in PBS for 1 h. This was followed by incubation in goat polyclonal antibody against microtubule-associated protein 1 light chain 3 (LC3; 1:100, sc-16756; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 24 h at 4 ℃ in a humidified chamber. After 3 washes for 10 min in PBS, the cells were incubated in donkey antigoat immunoglobulin G–fluorescein-isothiocyanate (1:400, sc-2024; Santa Cruz, USA) for 1 h at 4 ℃. Finally, cells were rinsed in PBS and examined with a fluorescence microscope (Eclipse TE 300, Nikon, Japan).
Cytofluorometric assessment of mitochondrial membrane potential To measure the mitochondrial membrane potential (ΔΨm), 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1), a sensitive fluorescent probe for ΔΨm was used[27]. The HL60 cells were cultured in 24-well plates and treated as follows: control group, 3-MA group, As2O3 group, As2O3+3-MA (pretreatment) group, and As2O3+3-MA (post-treatment) group. After exposure to the As2O3 (10 µmol/L) for 24 h, the cells were harvested and stained with 1 mL 10% RPMI-1640 medium containing 5 μmol/L JC-1 (Molecular Probes, USA) for 30 min at 37 ℃. Then the cells were rinsed with ice-cold PBS twice, resuspended in 1 mL ice-cool PBS, and immediately assessed for red and green fluorescence with flow cytometry[28].
JC-1 has been reported to differentially label mitochondria with high and low membrane potential[27]. JC-1 exists as a monomer and emits green fluorescence (λ=527 nm) at low membrane potential. However, at higher potentials, JC-1 forms “J-aggregates”, which emit red fluorescence (λ=590 nm). For the assessment of individual samples for JC-1 staining, a total of 10000 gated events were analyzed per sample using a flow cytometer (EPICS-XL, Beckman, USA). A 488 nm filter was used for the excitation of JC-1. Emission filters of 535 and 595 nm were used to quantify the population of mitochondria with green (JC-1 monomers) and red (JC-1 aggregates) fluorescence, respectively[28]. Frequency plots were prepared for FL1 (green) and FL2 (red) to determine the percentage of the mitochondria stained green (low membrane potential) and red (normal membrane potential).
Subcellular fractionations The HL60 cells were cultured in 60 mm dishes and divided into 5 treatment groups, as described earlier. After exposure to As2O3 (10 µmol/L) for 24 h, the cells were harvested and rinsed with ice-cold PBS twice. The isolation of mitochondria was performed as described previously[29]. The cells were suspended in buffer A (250 mmol/L sucrose, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 50 mmol/L Tris-HCl, 1 mmol/L DL-dithiothreitol (DTT), 1 mmol/L Phenylmethanesulfonyl fluoride (PMSF), 1 mmol/L benzamidine, 0.28 U/mL apotinin, 50 µg/mL leupeptin, and 7 µg/mL pepstatin A, pH 7.4) and homogenated with a glass Pyrex microhomogenizer (30 strokes). The homogenate was centrifuged at 1000×g at 4 ℃ for 10 min, and the resultant supernatant was transferred to a new Eppendorf tube and centrifuged at 10 000×g at 4 ℃ for 20 min to obtain the mitochondrial pellet and supernatant. The supernatant was transferred to a new tube and centrifuged at 100000×g for 1 h at 4 ℃ to generate the cytosolic fraction. The mitochondrial pellet was washed 3 times in buffer B (250 mmol/L sucrose, 1 mmol/L EGTA, 10 mmol/L Tris-HCl, 1 mmol/L DTT, 1 mmol/L PMSF, 1 mmol/L benzamidine, 0.28 U/mL apotinin, 50 µg/mL leupeptin, and 7 µg/mL pepstatin A, pH 7.4), spun at 10000×g at 4 ℃ for 10 min, and then lysed in Western blot lysing buffer.
Protein preparation and immunoblotting The cells were harvested and rinsed with ice-cold PBS twice. Five volumes of Western blot lysing buffer for each volume of cell pellets was added and the mixture was sonicated on ice (1 s/mL per sonicate, resting 30 s between intervals, a total of 5 times). The sample was centrifuged at 10000×g at 4 ℃ for 10 min and the supernatant was preserved at -70oC. Before immunoblotting, the protein concentrations were determined with a BCA detection kit (Pierce, USA) and adjusted to equal concentrations across different samples. The proteins were separated by 10% or 12% SDS–PAGE gel, transferred to nitrocellulose membranes, and immunoblotted with a goat polyclonal antibody against cathepsin B (1:100; sc-6493; Santa Cruz, USA), a goat polyclonal antibody against cathepsin L (1:100; sc-6499; Santa Cruz, USA), a mouse monoclonal antibody against cytochrome c (cyto-c) 1:1000; 7H8.2C12, PharMingen, San Diego, CA, USA) or rabbit polyclonal antibody against caspase-3 (1:200; H277; Santa Cruz, USA) at 4 ℃ overnight. The reactions of the primary antibodies were detected using horseradish peroxidase-conjugated antigoat, antimouse, or antirabbit antibodies (Sigma, USA) and were used at a 1:5000 dilution in blocking solution for 1 h at room temperature. Immunoreactivity was detected by enhanced chemiluminescence (ECL kit; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and visualized by autoradiography. Protein β-actin (1:5000; A5441; Sigma, USA) and heat shock protein 60 (HSP-60) (1:2000; H3524; Sigma, USA) were used as the loading controls.
Statistical analysis All data are presented as mean±SD. The statistical analysis was carried out by ANOVA followed by a Student’s t-test with P<0.05 representing significance.
Result
Increased AV following As2O3 treatment As2O3 inhibited the viability of the HL60 cells in a time- and dose-dependent fashion. After treatment with 20 µmol/L As2O3 for 12, 24, 48, and 72 h, the viability of the HL60 cells was significantly decreased (P<0.05). The rate of the inhibition of the viability of HL60 was 47.09%±2.01% (P<0.05) at 24 h, 72.18%±1.23% at 48 h, and 93.13%±0.04% (P<0.05) at 72 h, respectively, after As2O3 (10 μmol/L) treatment (results not shown).
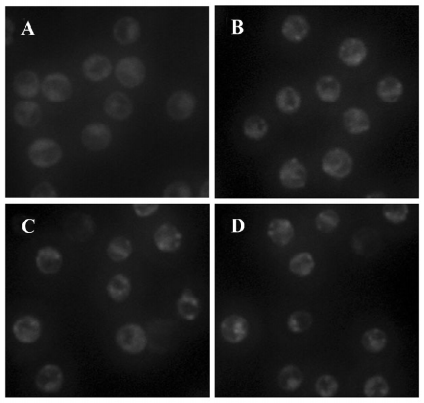
The autofluorescent substance MDC has been recently shown to be a marker for AV. When the cells were viewed with a fluorescence microscope, the AV labeled by MDC appeared as distinct dot-like structures distributed in the cytoplasm or in the perinuclear regions. We found that there was an increase in the number of MDC-labeled vesicles starting at 3 h and peaking at 6–12 h after As2O3 treatment, suggesting an induction of AV formation by As2O3 (Figure 1).
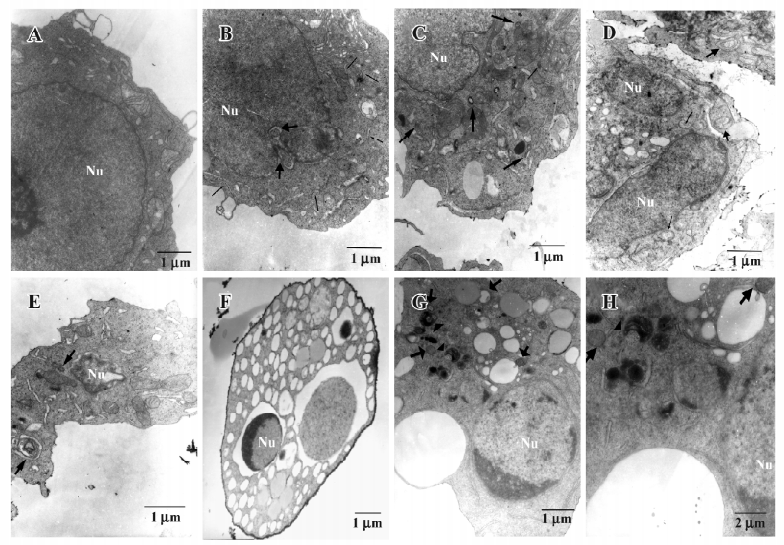
Apoptosis and autophagy are induced in HL60 cells exposed to As2O3 The ultrastructural analysis of the control cells showed a round shape and contained normal looking organelles, nuclei, and chromatin. While after As2O3 treatment, cells were found to contain many vesicles with the typical morphological features of autophagosomes, a number of isolated double-membrane structures were observed in the cytoplasm. These membrane structures engulfed cytoplasm fractions and organelles to form double or multimembrane autophagosomes. Mitochondria were swollen and lysosomal staining darkened, indicating the injury of mitochondria and the activation of lysosomes. Meanwhile, the morphological features of apoptosis, such as chromatin condensation and margination, were also observed (Figure 2).
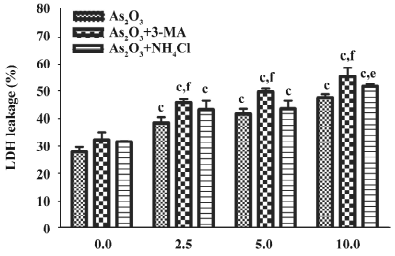
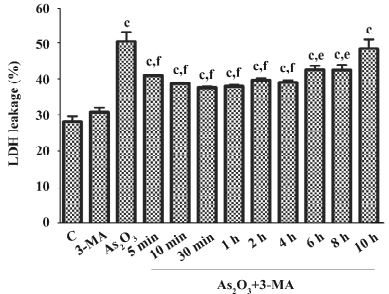
Effects of 3-MA and NH4Cl on the cytotoxicity of As2O3in HL60 cells When the specific autophagy inhibitor 3-MA or the lysosome-neutralizing agent NH4Cl were added 1 h before As2O3, LDH leakage significantly increased, suggesting that autophagy inhibits the cytotoxicity of As2O3 in HL60 cells (Figure 3). In contrast, after the treatment of HL60 cells with 3-MA 30 min after As2O3 (10 µmol/L), LDH leakage significantly decreased. The inhibitory effects of 3-MA on LDH leakage were observed when it was added to the culture 5 min–12 h after As2O3, suggesting that persistent autophagy activation enhances the cytotoxicity of As2O3 in HL60 cells (Figure 4).
LC3 activity assay LC3 is considered as a marker of cellular autophagosomes, and was observed as green punctuated labeling distributed in the cytoplasm after treatment with As2O3 (10 µmol/L). The negative control cells only showed diffuse immunostaining; pretreatment with the lysosome-neutralizing agent NH4Cl attenuated the immunoreactitvity of LC3 induced by As2O3; treatment with the specific autophagy inhibitor 3-MA before or after As2O3 inhibited the immunoreactivity of LC3 completely (Figure 5).
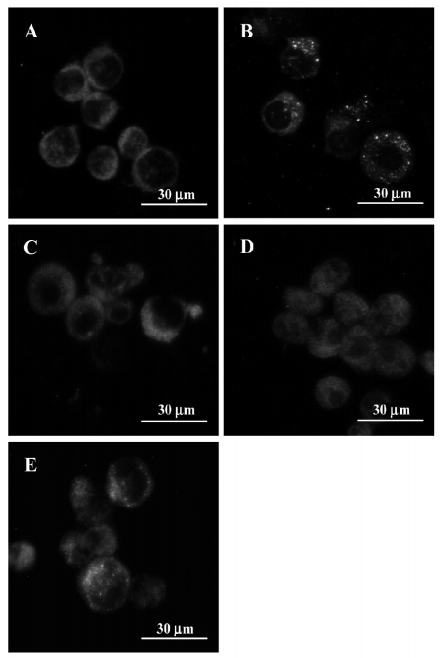
Effects of 3-MA on As2O3-induced activation of cathepsins B and L The Western blot analysis revealed that As2O3 increased the protein levels of cathepsins B and L. 3-MA alone had no effect on the protein levels of cathepsins B and L. Post-treatment with 3-MA 30 min after As2O3 (10 µmol/L) lessened As2O3-induced increases in cathepsin L protein levels, while pretreatment with 3-MA 1 h before As2O3 enhanced As2O3-induced increases in cathepsin L protein levels. The same trend was seen on As2O3-induced increases in cathepsin B, although it was less robust (Figure 6).
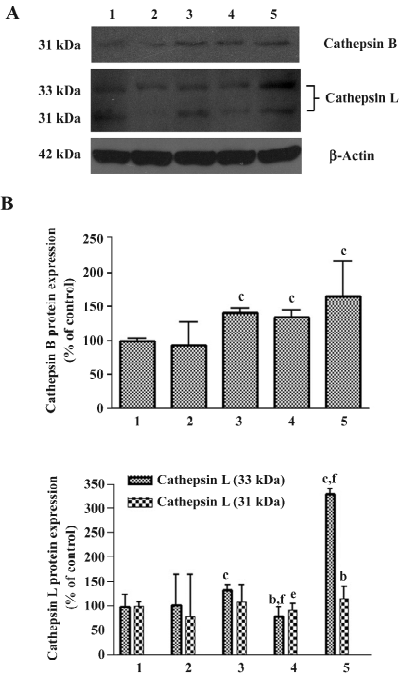
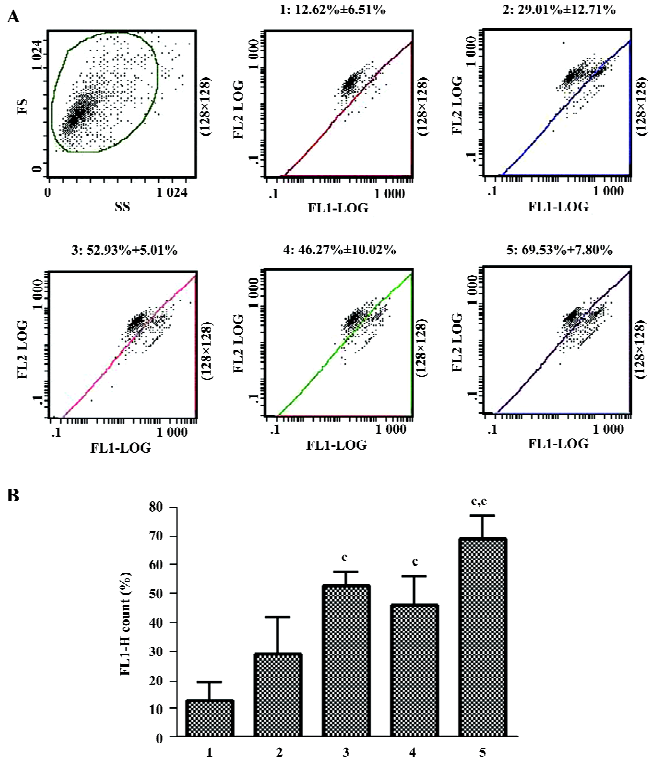
Effects of 3-MA on As2O3-induced ΔΨm collapse When ΔΨm was low, JC-1 existed mainly in a monomeric form, which emits green fluorescence. As shown in Figure 7, after exposure to As2O3 for 24 h, an increase in the population of mitochondria with collapsed membrane potential was observed. When autophagy was inhibited with 3-MA 30 min after As2O3 treatment, the decrease in ΔΨm induced by As2O3 was slightly attenuated, but it had no significant difference compared to As2O3 alone. However, when autophagy was blocked 1 h before As2O3 treatment, the collapse of ΔΨm induced by As2O3 was significantly exacerbated (Figure 7).
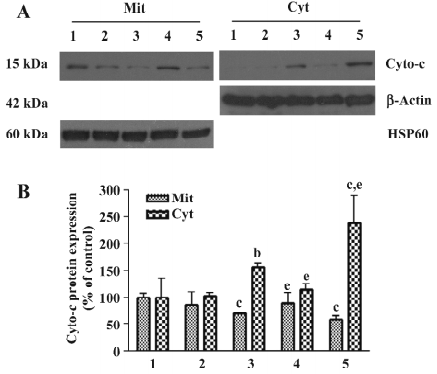
Effects of 3-MA on As2O3-induced cyto-c redistribution and caspase-3 activation Cyto-c release from mitochondria into the cytoplasm is a critical event in apoptosis. The Western blot analysis results demonstrated that the release of cyto-c took place when the HL60 cells were exposed to As2O3 for 24 h. As2O3-induced cyto-c release was significantly inhibited by post-treatment with 3-MA 30 min after As2O3 treatment. In contrast, As2O3-induced cyto-c release was enhanced by pretreatment with 3-MA 1 h before As2O3 (Figure 8).
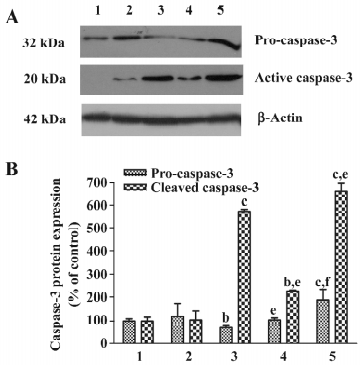
The redistribution of cyto-c could initiate a cascade of caspase activation. As seen in Figure 9, after treatment with As2O3 for 24 h, caspase-3 was activated, as revealed by the production of 20 kDa active caspase fragments. The blockade of autophagy 30 min after As2O3 treatment with 3-MA significantly suppressed the As2O3-induced activation of caspase-3, while blockage of autophagy with 3-MA 1 h before As2O3 treatment potentiated the As2O3-induced activation of caspase-3 (Figure 9).
Discussion
Extensive research has revealed that As2O3 exerts potent antitumor effects against several types of cancers, including leukemia[17,30-32], and it has been recognized as being highly effective in clinics for the treatment of APL[33,34]. It is believed that the antitumor efficacy of As2O3 involves several actions, including the inhibition of proliferation, induction of incomplete differentiation, and promotion of apoptosis of tumor cells[35–37], but the underlying molecular mechanisms of its antitumor effects still remain elusive. Autophagy is a gatekeeping mechanism for stabilizing cell homeostasis[38]. Studies have shown that autophagy plays important roles in physiology and pathophysiology[39]. There is a potential link between autophagy and a number of human diseases, such as cancer, cardiomyopathy, and neurodegenerative disorders, including Alzheimer’s, Parkinson’s, and Huntington’s[40]. Therefore, the study of the involvement of autophagy in the As2O3-induced death of HL60 cells would result in a better understanding of the molecular mechanisms by which As2O3 inhibits tumor cells and would also result in proposals for new therapeutic approaches for leukemia.
In the present study, it was found that the viability of HL60 cells was significantly inhibited by As2O3 and vacuoles labeled by MDC increased in the cytoplasm, suggesting the induction of autophagy in HL60 cells after As2O3 administration. At present, transmission electron microscopic examination is still the most reliable means to detect the induction of autophagy and the dynamic morphological changes of autophagosomes and autolysosomes. After exposure to As2O3 for 3 h, a number of isolated double-membrane structures and autophagosomes could be identified in HL60 cells with TEM. When the duration of As2O3 treatment was prolonged to 24 h, coexistence of apoptotic features, such as cell shrinkage, chromatin condensation, and margination were observed.
LC3 is an autophagosomal ortholog of yeast Atg8, and is considered an autophagosomal marker. LC3 modification is essential for the autophagic process. When the autophagosomes were induced, LC3 was cleaved to a smaller protein (LC3-II). Cleaved LC3 is redistributed from the cytoplasm to the membranes of pre-autophagosomes and autophagosomes, thus the pattern of immunostaining of LC3 will be altered from a diffuse to punctate pattern[41]. After treatment with As2O3, we observed green punctate immunostaining of LC3 scattered in the cytoplasm, suggesting that As2O3 activated autophagy. 3-MA is a class III phosphatidylinositol 3-kinase inhibitor[42]. Early in 1982, it was found that 3-MA inhibits the formation of autophagosomes[43], and now it is generally accepted as a specific inhibitor of autophagy. In our study, it was shown that pre- or post-treatment with 3-MA completely inhibited the activation of LC3 induced by As2O3, indicating that autophagy maturation was fully suppressed by 3-MA. The lysosome is an important organelle in autophagy. In the last step of autophagy, the autophagosome targets the lysosome, where its outer membrane fuses with the lysosomal membrane; the inter sac (autophagic body) enters the lysosome/vacuole and is then degraded in the lysosome/vacuole so the carrying constituent components can be recycled[1]. As shown in the results of immunofluorescence, lysosome-neutralizing agent NH4Cl attenuating the expression of LC3 induced by As2O3 due to injury to the lysosomal function, suggesting it could partly inhibit autophagy.
To determine the contribution of autophagy to the As2O3-induced death of HL60 cells, the present study examined the effects of the inhibition of autophagy on the cytotoxicity of As2O3. In this work, 10 mmol/L 3-MA (an effective dose for the inhibition of autophagy, as reported by many investigators) was shown to potentiate the cytotoxicity As2O3 in HL60 cells when it was administered 1 h before As2O3. The present study also found that the same dose of 3-MA weakened the cytotoxicity of As2O3 when it was administered after As2O3 (10 μmol/L) treatment. These data raise the possibility that autophagy might have dynamic effects on the As2O3-induced death of HL60 cells. In the early stage, autophagy can be a survival pathway representing a defense mechanism against As2O3-induced toxicity in HL60 cells, and in the later stage, autophagy can also be a death pathway leading HL60 cells to apoptotic or autophagic cell death.
Recently, the role of autophagy in cell survival and death has become an important subject of research. However, the significance of autophagy in various diseases remains to be determined[40,44–46]. In our previous study for a role of autophagy in the snake venom crotoxin-induced death of tumor cell lines, we found that autophagy played different roles in the crotoxin-induced death of K562 cells and MCF-7 cells. In K562 cells, autophagy delayed crotoxin-induced cell death, while in MCF-7 cells, which are deficient in caspase-3 activity, autophagy promoted crotoxin-induced cell death[19,20]. The present findings further suggest that autophagy has different effects on cell survival depending on the stages of cell death process. Autophagy activation shortly after exposure of HL60 cells to As2O3 exerted protective effects. However, persistent activation or overactivation of autophagy exerted destructive effects. This raises the possibility that in the early stage, when the death signals induced by As2O3 were relatively weak, autophagy is activated as an adaptive response to maintain HL60 cell survival under stress conditions via increasing protein catabolism; the elimination of excess or damaged organelles like peroxisomes, mitochondria, and the endoplasmic reticulum; and sequestering noxious substances, thus delaying the induction of apoptosis. In contrast, in the late stage, with the continuous presence of As2O3, cell death signals are reinforced to cause the aggravated damage of HL60 cells, and then the defense mechanism of autophagy ultimately fails in its mission to preserve cell viability and turns to induce an autophagic cell death or promote the apoptotic cascade.
There is complex interplay between autophagic and apoptotic pathways, but the details of its mechanisms and molecular switching points between these 2 pathways in tumor cells have still not been fully elucidated. It is speculated that mitochondria may be the critical organelle that integrate the 2 types of cell death. In the apoptotic process, the mitochondrial permeability transition (MPT) would initiate the release of mitochondrial proapototic proteins, such as cyto-c and apoptosis-inducing factor, from the intermembrane space. These proteins activate a family of proteolytic enzymes called caspases and other downstream events in apoptotic cell death[47–49]. Now it has been proposed that MPT might account for the induction of autophagy[50]. Studies have revealed that a good gatekeeper protein of autophagy, the mammalian target of rapamycin, existed in mitochondria, and the damaged mitochondria could initiate autophagy[51–53]. Damaged mitochondria could be selectively sequestered into autophagosomes and delivered to lysosomes for degradation[54]. The genetic data from mammals revealed that the deletion of autophage-specific gene Atg7 resulted in the accumulation of mitochondria with abnormal morphology[55]. In conclusion, apoptosis and autophagy may be the different results caused by the unified regulation of mitochondria[56,57]. In the present study, we found that following treatment with As2O3, ΔΨm collapsed, cyto-c was released from mitochondria to the cytoplasm, and caspase-3 was activated. Moreover, pretreatment with the autophagy-specific inhibitor 3-MA prior to As2O3 amplified the above-mentioned apoptotic signaling pathways, while it inhibited these signaling pathways when 3-MA was administered post-As2O3 insult. We speculate that in the earlier period, autophagy induced by As2O3 could delete impaired mitochondria and sequester certain proapoptotic molecules, such as cyto-c, resulting in the blockage or retardation of apoptosis. However, in the late period, As2O3-induced autophagy could cause autophagic death or amplification of the mitochondria-mediated apoptotic signaling pathway possibly through the lysosomal pathway.
Another organelle that plays a causal role in both autophagy and apoptosis is the lysosome, a major organelle where most proteins are broken down by proteinases[1,58]. There are many different hydrolases in lysosomes[59-61]. Among them, cysteine proteinases such as cathepsins B and L are the major lysosomal proteases, but their exact roles are still unknown. A wide variety of death stimuli can induce partial lysosomal membrane permeabilization and release cathepsins into the cytosol. The leakage of active cathepsins triggers caspase- and mitochondrion-independent programmed cell death[62–65]. It has also been documented that cathepsins could activate the classic apoptosis pathway possibly by cleaving the proapoptotic Bcl-2 family member Bid[66]. Autophagic cargos, including cytoplasmic soluble constituents and cellular organelles (such as mitochondria and peroxisomes) engulfed by autophagosomes would eventually be delivered to the lysosomal lumen and degraded by numerous hydrolases, while leupeptin, a potent inhibitor of cysteine proteinases, has been shown to increase the density of lysosomes due to the accumulation of undergraded proteins sequestered by autophagy and/or heterophagy[67–69]. In this study, we found that cathepsins B and L were activated after exposure to As2O3. Pretreatment with 3-MA before As2O3 increased the expression of cathepsin L, while post-treatment with 3-MA after As2O3 depressed it. We speculate that cathepsins, especially cathepsin L, might also be involved in the interaction of autophagy and apoptosis.
In summary, this study suggests that autophagy and apoptosis could be triggered by As2O3 in HL60 cells. The data also suggest that autophagy has differential effects on the As2O3-induced death of HL60 cells: it may be a protective mechanism against apoptosis in the earlier period of As2O3 treatment, while it promotes apoptosis and/or leads to autophagic death in the later period of As2O3 treatment. Autophagy regulated cell survival may through interfering with the mitochondria-mediated apoptotic pathway.
References
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun 2004;313:453-8.
- Bursch W, Ellinger A, Gerner C, Schulte-Hermann R. Autopha-gocytosis and programmed cell death. In: Klionsky DJ, editors. Autophagy. Landes Bioscience, Georgetown, 2004. p 287–303.
- Lockshin RA, Osborne B, Zakeri Z. Cell death in the third millennium. Cell Death Differ 2000;7:2-7.
- Lockshin RA, Zakeri Z. Apoptosis, autophagy and more. Inter J Biochem Cell Biol 2004;36:2405-19.
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004;23:2891-906.
- Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ, et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol 2003;13:358-63.
- Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol 2003;13:350-7.
- Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res 2001;268:139-49.
- Knaapen MW, Davies MJ, De-Bie M, Haven AJ, Martinet W, Kockx MM. Apoptotic versus autophagic cell death in heart failure. Cardiovasc Res 2001;51:304-12.
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 2004;16:663-9.
- Lockshin RA, Zakeri Z. Caspase-independent cell deaths. Curr Opin Cell Biol 2002;14:727-33.
- Knecht E, Hernández-Yago J, Grisolía S. Regulation of lysosomal autophagy in transformed and non-transformed mouse fibroblasts under several growth conditions. Exp Cell Res 1984;154:224-32.
- Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE, Baccino FM, et al. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis 1993;14:2501-5.
- Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997;89:3345-53.
- Roboz GJ, Dias S, Lam G, Lane WJ, Soignet SL, Warrell RP Jr, et al. Arsenic trioxide induces dose- and time-dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood 2000;96:1525-30.
- Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 2003;63:2103-8.
- Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005;24:980-91.
- Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res 2007;31:329-39.
- Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P, Qin ZH. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006;47:521-30.
- Yan CH, Qin ZH, Yang YP, Gu ZL, Liang ZQ. Involvement of autophagy and caspase-independent apoptosis in crotoxin-induced death of MCF-7 cells. Acta Pharmacol Sin 2007;28:540-8.
- Vistica DT, Skehan P, Scudiero D, Monks A, Pittman A, Boyd MR. Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res 1991;51:2515-20.
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 1995;66:3-14.
- Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 2002;3:472-82.
- Thomas CE, Mayle DA. NMDA-sensitive neurons profoundly influence delayed staurosporine-induced apoptosis in rat mixed cortical neuronal cultures. Brain Res 2000;884:163-73.
- Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci 1997;9:159-69.
- Hong QT, Song YT, Tang YP, Liu CM. Determination and application of leakage rate of lactate dehydrogenase in the cultured medium of cells. Chin J Cell Biol 2004;26:89-92.
- Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess DY changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 1997;411:77-82.
- Gravance CG, Garner DL, Baumber J, Ball BA. Assessment of equine sperm mitochondrial function using JC-1. Theriogenology 2000;53:1691-703.
- Qin ZH, Wang YM, Kikly KK, Sapp E, Kegel KB, Aronin N, et al. Pro-caspase-8 is predominantly localized in mitochondia and released into cytoplasm upon apoptotic stimulation. J Biol Chem 2001;276:8079-86.
- Akao Y, Nakagawa Y, Akiyama K. Arsenic trioxide induces apoptosis in neuroblastoma cell lines through the activation of caspase 3 in vitro. FEBS Lett 1999;455:59-62.
- Chow SK, Chan JY, Fung KP. Inhibition of cell proliferation and the action mechanisms of arsenic trioxide (As2O3) on human breast cancer cells. J Cell Biochem 2004;93:173-87.
- Yi J, Gao F, Shi G, Li H, Wang Z, Shi X, et al. The inherent cellular level of reactive oxygen species: One of the mechanisms determining apoptotic susceptibility of leukemic cells to arsenic trioxide. Apoptosis 2002;7:209-15.
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997;89:3354-60.
- Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti L, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 1998;339:1341-8.
- Park JW, Choi YJ, Jang MA, Baek SH, Lim JH, Passaniti T, et al. Arsenic trioxide induces G2/M growth arrest and apoptosis after caspase-3 activation and bcl-2 phosphorylation in prominute socytic U937 cells. Biochem Biophys Res Commun 2001;286:726-34.
- Zhu Q, Zhang JW, Zhu HQ, Shen YL, Flexor M, Jia PM, et al. Synergic effects of arsenic trioxide and Camp during acute promyelocytic leukemia cell maturation subtends a novel signaling cross-talk. Blood 2002;99:1014-22.
- Jiang XH, Wong BC, Yuen ST, Jiang SH, Cho CH, Lai KC, et al. Arsenic trioxide induce s apoptosis in human ga stric cancer cells through up-regulation of p53 and activation of caspase-3. Int J Cancer 2001;91:173-9.
- Autophagy Kelekar A. Ann N Y Acad Sci 2005;1066:259-71.
- Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 2006;27:411-25.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004;306:990-5.
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19:5720-8.
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3' -kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000;275:992-8.
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982;79:1889-92.
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005;115:2679-88.
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005;1:11-22.
- Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003;1603:113-28.
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000;6:513-9.
- Saikumar P, Dong Z, Mikhailov V, Denton M, Weinberg JM, Venkatachalam MA. Apoptosis definition, mechanism, and relevance to disease. Am J Med 1999;107:489-506.
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annau Rev Cell Dev Biol 1999;15:269-90.
- Mijaljica D, Prescott M, Devenish RJ. Different fates of mitochondria: alternative ways for degradation? Autophagy 2007;3:4-9.
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001;15:2286-7.
- Cardenas ME, Cutler NS, Lorenz MC, Di-Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 1999;13:3271-9.
- Levine B, Klionsky DJ. Development by Self-Digestion:Molecular Mechanisms and Biological Functions of Autophagy. Dev Cell 2004;6:463-77.
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3-5.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005;169:425-34.
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366:177-96.
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 2000;14:729-39.
- Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene 2004;23:2881-90.
- Barrett AJ, Kirschke H. Cathepsin B, Cathepsin H, and cathepsin L. Methods Enzymol 1981;80:535-61.
- Takahashi T, Tang J. Cathepsin D from porcine and bovine spleen. Methods Enzymol 1981;80:565-81.
- Nikawa T, Towatari T, Katunuma N. Purification and characterization of cathepsin J from rat liver. Eur J Biochem 1992;204:381-93.
- Nakamura Y, Takeda M, Suzuki H, Morita H, Tada K, Hariguchi S, et al. Lysosome instability in aged rat brain. Neurosci Lett 1989;97:215-20.
- Bi X, Yong AP, Zhou J, Gall CM, Lynch G. Regionally selective changes in brain lysosomes occur in the transition from young adulthood to middle age in rats. Neuroscience 2000;97:395-404.
- Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. Int J Biochem Cell Biol 2005;37:2098-114.
- Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene 2004;23:2746-56.
- Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM, et al. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 2004;279:3578-87.
- Furuno K, Ishikawa T, Kato K. Appearance of autolysosomes in rat liver after leupeptin treatment. J Biochem 1982;91:1485-94.
- Kominami E, Hashida S, Khairallah EA, Katunuma N. Sequestration of cytoplasmic enzymes in an autophagic vacuole-lysosomal system induced by injection of leupeptin. J Biol Chem 1983;258:6093-100.
- Ohshita T, Kominami E, Ii K, Katunuma N. Effect of starvation and refeeding on autophagy and heterophagy in rat liver. J Biochem 1986;100:623-32.