
Synergistic actions of diacylglycerol and inositol 1,4,5 trisphosphate for Ca2+-dependent inactivation of TRPC7 channel1
Introduction
Transient receptor potential (TRP) is a vast gene superfamily which contains more than 30 isoforms ranging from yeast to mammals[1–3]. The proteins of this family are predicted to have 6 transmembrane (TM) domains and intracellularly located amino and carboxyl termini. Like many voltage-gated channels, a lipophilic hairpin structure between the fifth and the sixth TM domain is predicted to form the channel pore[4]. Functional studies in vitro show that most channels of this family have poor selectivity over cations and can be activated through G protein-coupled receptors or tyrosine kinase receptors linked to phospholipase C[4].
The mammalian TRP superfamily is composed of 6 subfamilies: the canonical TRP (TRPC), the vanilloid receptor TRP (TRPV), the melastatin or long TRP, the mucolipins, the polycystins, and ankyrin transmembrane protein 1[3]. Among these, TRPCs share the highest similarities to its prototype, Drosophila TRPs, and are likely to be the molecular candidate that mediate the native receptor-operated and store-operated Ca2+ entry observed in many non-excitable tissues[1–3]. The 7 members in TRPC can be subclassified into 3 subgroups according to the structural and functional similarities: TRPC3/6/7, TRPC1/4/5, and TRPC2[1-3]. TRPC7 is the latest cloned TRPC isoform and least elucidated for its channel regulation.
Ca2+ entry through the channel pore plays essential roles in the regulation of many Ca2+ permeable channels. It is well known that the L-type voltage-gated Ca2+ current decays much faster in the presence of extracellular Ca2+ than its surrogate Ba2+ [5]. The ubiquitous Ca2+ sensor calmodulin (CaM) tethered to the proximal segment of the cytoplasmic C-terminus of the a1c subunit, upon entry of Ca2+, is thought to reorientate to cause conformational rearrangements[6–8]. TRPV1 is probably the best-defined channel in the TRP superfamily that serves as a poly-modal detector of many pain-producing chemicals and physical stimuli in the dorsal root ganglia neurons[9,10]. This non-selective cationic channel undergoes rapid inactivation or desensitization during the agonist application in Ca2+-containing bathing solution[11], but the underlying mechanism is controversial. While Numazaki et al[12] pointed out that Ca2+/CaM binded to a 35 aa segment of the C-terminus of the channel, resulting in desensitization, the involvement of calcineurin-dependent dephosphorylation was long regarded by others to be involved in the process[13]. In addition, Bhave et al[14] showed that cAMP dependent phosphorylation antagonizes the desensitization leading to the resensitization of the channel.
TRPC7 is highly sensitive to extracellular Ca2+ (Ca2+o) in the physiological range, being almost completely inhibited by 1 mmol/L Ca2+o [IC50 (half inhibition-concentration value)=0.11 mmol/L][15]. A detailed examination with single channel recordings revealed that rapid permeation blockade by external Ca2+ and the tonic inhibitory actions of internal Ca2+ via Ca2+/CaM both underlie this inhibition. According to our preliminary results, the inhibition of TRPC7 induced by the transient application of 1 mmol/L Ca2+o was hardly washable, suggesting that some long-term effects of Ca2+o still function in the channel inactivation regulation. To explore the mechanisms underlying this long-term effect and to broaden our understanding towards this highly Ca2+-sensitive TRPC7 channel, we performed further patch-clamp experiments in the present study.
Material and methods
Cell culture and transfection Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. For transfection, the cells were reseeded in a 35 mm culture dish and were allowed to grow to a 40%–50% confluency. The cells were then transfected with a mixture of 2 µg plasmid vector (pCI-neo) incorporating murine TRPC7 DNA and 0.4 µg pCI-neo-πH3-CD8 (cDNA of the T cell antigen CD8) with the aid of 20 µL of the transfection reagent SuperFect (Qiagen, Germany). In some cases (Figure 1F), 2 µg plasmid DNA for a Ca2+-insensitive mutant calmodulin (replacement with alanine of the aspartate residues 21, 57, 94, and 130 in 4 E–F hands of calmudulin) was cotransfected. Approximately 24 h later, the cells were trypsinized and reseeded onto coverslips precoated with 100 µmol/L poly-L-lysine. Electrophysiological experiments were performed within 24–48 h.
Electrophysiological recording Two modes of patch-clamp recording technique were employed in the present study, that is, nystatin-perforated and conventional whole-cell recording. For both recordings, microglass pipettes with 2.5–4.0 ΜΩ resistance (when filled with Cs-pipette solution) were used. Voltage generation and current signal acquisition were implemented by a high-impedance, low-noise patch-clamp amplifier (EPC9; HEKA Electronics, Lambrecht/Pflaz Germany). For the construction of the current–voltage curve (Figure 1B), sampled data were low-pass filtered at 1 kHz and stored on a computer hard disc after digitization at 5 kHz. Long-term recordings (current traces, such as Figure 1A) were performed in conjunction with an A/D, D/A converter, Powerlab/400 (AD instruments, Bella Vista, New South Wales, Australia) at a sampling rate of 100 Hz to diminish the size of the data; data evaluation was made with accessory software, Chart v3.6. All the drugs were administered by a “Y-tube” device that enabled us to quickly change the solution around the cell.
Solutions Solutions with the following composition were used. The pipette solution for the nystatin-perforated recording (in mmol/L) was as follows: 140 CsCl, 2 MgCl2, 10 HEPES (4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid), and 10 glucose (adjusted to pH 7.2 with Tris base); the poorly-buffered pipette solution for conventional whole-cell recording (in mmol/L) was: 120 CsOH, 120 aspartate, 20 CsCl, 2 MgCl2, 0.1 BAPTA (1,2 bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid) or EGTA (ethylene glycol-bisc2-aminoethylether)-N,N,N’,N’-tetraacetic acid), 0.04 CaCl2, 10 HEPES, 2 ATP, 0.1 GTP (guanosine 5’-triphosphate), and 10 glucose (adjusted to pH 7.2 with Tris base); the highly-buffered pipette solution for conventional whole-cell recording (in mmol/L)was: 120 CsOH, 120 aspartate, 20 CsCl, 2 MgCl2, 10 BAPTA (or EGTA), 4 CaCl2, 10 HEPES, 2 ATP, 0.1 GTP, and 10 glucose (adjusted to pH 7.2 with Tris base). In total, 100 μmol/L GTPγS, 100 μmol/L inositol 1,4,5 trisphosphate (IP3), inositol 1,3,4,5 tetraphosphate (IP4), or 1,4 bisphosphate (IP2) was added to the poorly-buffered or highly-buffered pipette solutions when necessary. The normal external solution contained (in mmol/L) 140 NaCl, 5 KCl, 1.2 MgCl2, 1 EGTA, 10 HEPES, and 10 glucose (adjusted to pH 7.4 with Tris base). For the Ca2+-containing external solution, 1 mmol/L CaCl2 was added with the omission of EGTA.
Drugs 1-oleoyl-2-acetyl-sn-glycerol (OAG), GTPγS, heparin, thapsigargin (TG), and protein kinase C (PKC) inhibitory peptide (PKC–IP19–36) were purchased from Calbiochem (La Jolla, CA, USA); IP2, IP4, carbachol (CCh), and HEPES were from Sigma (St Louis, MO, USA); ATP, GTP, EGTA, and BAPTA were from Dojindo (Kumamoto, Japan).
Statistics All data are expressed as means±SEM. Paired and unpaired Student’s t-test was used to evaluate statistical significance.
Results
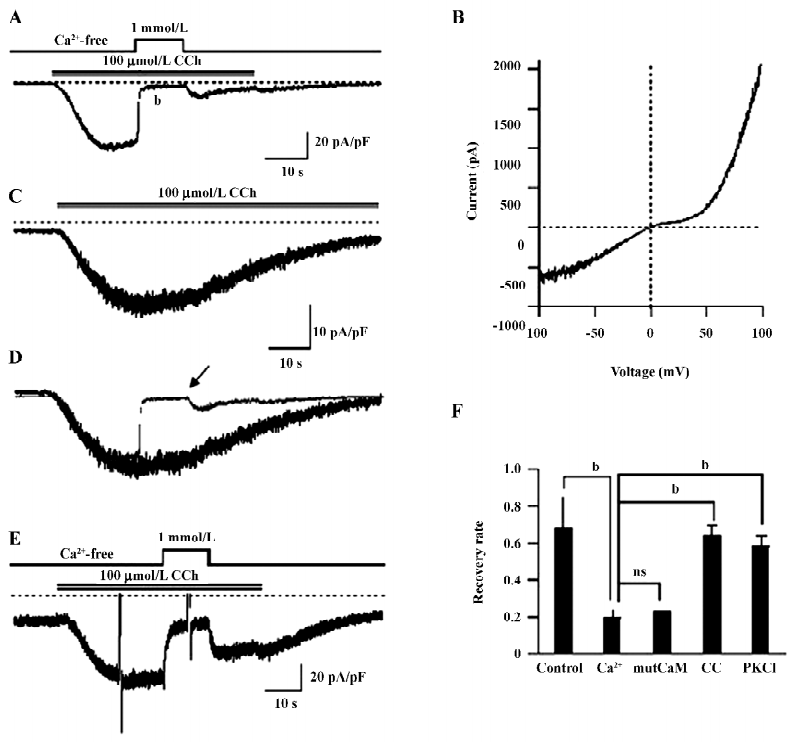
Ca2+-dependent inactivation of TRPC7 current HEK293 cells overexpressing mouse TRPC7 exhibited large inward currents following the addition of 100 µmol/L CCh (35.7±5.2 pA/pF, n=21; Figure 1A). The current–voltage (I-V) curve showed a double-rectifying property with a reversal potential near 0 (Figure 1B), which represents the typical I–V relationship of the TRPC3/6/7 subfamily[1]. Little current was recorded with empty vector-expressing cells (0.89±0.61 pA/pF, n=10). Thus, the currents induced by CCh in the present study resulted from the heterologously-expressed TRPC7 channel (designated as ITRPC7 hereafter).
The application of 1 mmol/L Ca2+o after full activation by CCh caused a rapid decline of ITRPC7 (Figure 1A). According to Shi et al[15], this inhibition can be assigned to the blockade of cation permeation by external Ca2+, which accounts for approximately 80% of the observed inhibition and the intracellular Ca2+/CaM-dependent tonic inhibition, which accounts for the remaining 20%. The inhibition of ITRPC7 by Ca2+o was long lasting since only a small fraction of the current was recovered after Ca2+o was washed out (Figure 1A). Superimposition of traces with (Figure 1A; indicated by an arrow) and without (Figure 1C) transient Ca2+o application clearly demonstrates that Ca2+o greatly accelerates the inactivation process of ITRPC7 (Figure 1D). As summarized in Figure 1F, the recovery rate of ITRPC7 (the ratio of current amplitudes after to before Ca2+ application; in the control, this ratio was calculated as the relative amplitude of ITRPC7 10 s after the peak to the maximum) at the 2 conditions showed a statistically significant difference. The persistent inhibition of ITRPC7 by Ca2+o is unlikely due to the remaining function of intracellular CaM, since a similar recovery rate was observed when a Ca2+-insensitive mutant calmodulin (mutCaM) was co-expressed with TRPC7 (Figure 1F). Consistent with the previous report[15], however, PKC is rather involved in the process since both pretreatment of the cells with calphostin C (500 nmol/L), a potent inhibitor of PKC, and inclusion of PKC–IP19–36 (5 µmol/L) in the pipette solution antagonized the persistent Ca2+ inhibition and accordingly raised the recovery rate, respectively (Figure 1E,1F).
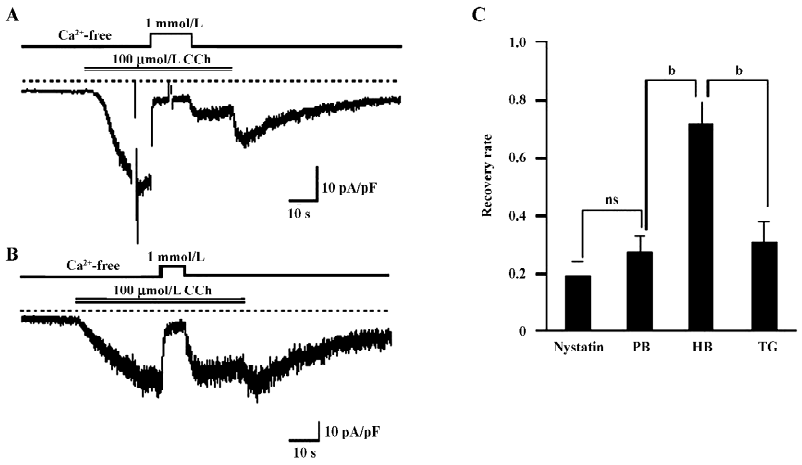
Ca2+ entry is involved in the Ca2+-dependent inactivation of TRPC7 To determine whether the Ca2+-dependent inactivation (CDI) of ITRPC7 is due to Ca2+ influx, we next employed 2 different pipette solutions with weak and high-buffering capacities for Ca2+. As shown in Figure 2A and 2B, when recorded with high-buffering pipette solution (10 mmol/L BAPTA plus 4 mmol/L Ca2+), a brief Ca2+ addition no longer facilitated the inactivation of ITRPC7. As summarized in Figure 2C, the recovery rate of ITRPC7 with weak Ca2+ buffering was similar to that with the nystatin-perforated recording, but was significantly smaller than that obtained under strong Ca2+-buffering conditions. These data preliminarily suggest that the source of Ca2+ that causes this persistent inhibition is due to Ca2+ entry because the Ca2+ buffer power in chelating the transient Ca2+ entry near the vicinity of the channel might be weak. To reinforce this idea, TG, a potent inhibitor of Ca2+-ATPase in the endoplasmic reticulum (ER), was used to deplete the internal Ca2+ store. As summarized in Figure 2C, the recovery rate of the Ca2+ inhibition of ITRPC7 did not change significantly when the bathing solution contained 2 µmol/L TG. These results collectively suggest that Ca2+ entry through the activated TRPC7 channel accelerates the inactivation of the channel.
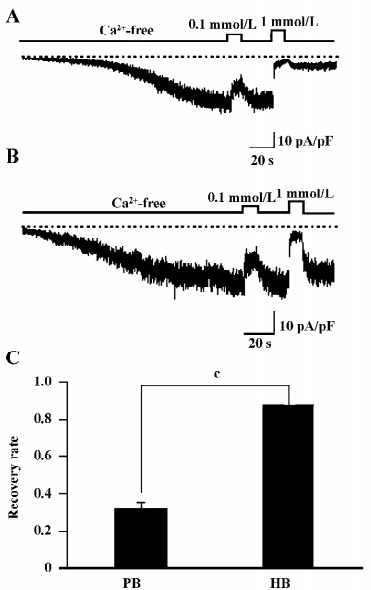
GTPγS-induced TRPC7 current shows similar CDI It is well established that Ca2+ affects the inactivation and/or desensitization of channels through acting on both the G protein-coupled receptor and its downstream effectors-mediated pathways[16-19]. To delineate the site of actions of Ca2+ on TRPC7, GTPγS (100 µmol/L) was included in the pipette solution. Dialysis of the cell with GTPγS resulted in the slow development of an inward current comparable to that evoked by CCh (Figures 3A,3B). The addition of Ca2+o caused a dose-dependent inhibition of the current. Perfusion of 0.1 mmol/L Ca2+o mildly inhibited the current, which was quickly reversed by the removal of Ca2+ (Figures 3A,3B). In contrast, with 1 mmol/L Ca2+o, which severely inhibited the current, the recovery from the inhibition became much less complete (Figure 3A,3C), and was rescued by the inclusion of high concentrations of BAPTA in the pipette (Figure 3B,3C). Because GTPγS directly activates the G protein and bypasses the receptor stimulation, these results suggest that the CDI of ITRPC7 does not involve a process upstream of G protein activation.
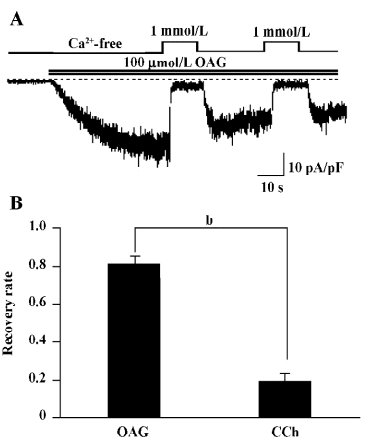
OAG-induced TRPC7 current exhibits no obvious CDI The members of the TRPC3/6/7 subfamily are known to be activated by 1 of the major phosphatidylinositide metabolites, diacylglycerol (DAG). This action is thought to be direct on the channel protein, independent of PKC or IP3[20]. Thus, the use of DAG instead of CCh may enable us to explore the above-mentioned actions of Ca2+ in simpler conditions in which the role of IP3 can be excluded. For this purpose, we used a membrane-permeable analog of DAG, OAG. Perfusion of 100 µmol/L OAG induced an inward current with the hallmarks of ITRPC7 (ie the reversal potential near 0; the ohmic current–voltage relationship with a slight double-rectifying property; blockade by millimolar extracellular Ca2+; data not shown). The sustained inhibition after application of 1 mmol/L Ca2+o seen for CCh- and GTPγS-induced ITRPC7 was almost completely abolished in OAG-induced ITRPC7 (Figure 4A). The recovery from the Ca2+-dependent inhibition was much more complete (80%) in OAG-induced ITRPC7 as compared with that observed with CCh (circa 20%; Figure 4B).
IP3 regresses the CDI of OAG induced currents The most important difference in activating the TRP channels between DAG and GTPγS/CCh is no involvement of the IP3 pathway in the former. Therefore, 100 µmol/L (the commonly-used full concentration) IP3 was added into the pipette solution to see whether the Ca2+-induced persisting inhibition of ITRPC7 could be restored. As illustrated in Figure 5A and summarized in Figure 5C, in more than half of the tested cells (6 of 11 cells), the intrapipette inclusion of IP3 caused a prominent, irreversible Ca2+ inhibition of ITRPC7 evoked by OAG. The effect of IP3 appeared to be direct since no such regressions of CDI were observed when other metabolites of inositol phosphate, such as IP4 and IP2, were included in the pipette solution (Figure 5B,5C).
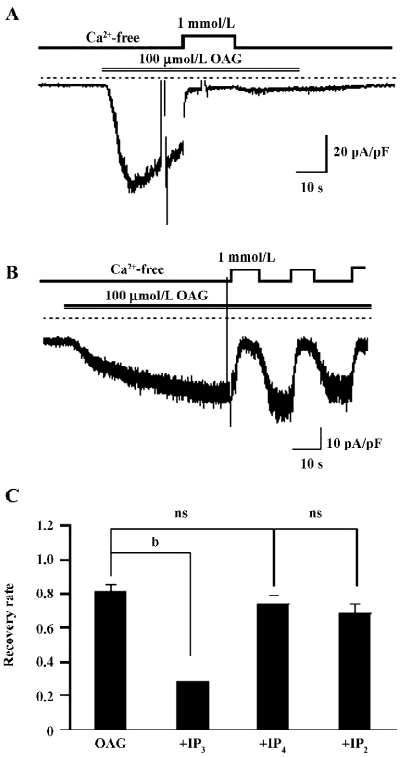
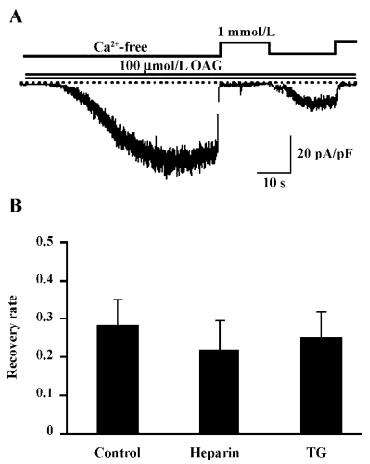
Effect of IP3 is IP3 receptor independent The main biological function of IP3 is to cause Ca2+ release through binding to the IP3 receptor (IP3R) on the ER. To examine whether the recovery of CDI caused by IP3 is due to the activation of IP3R, heparin, a potent IP3R antagonist (0.5 mg/mL), was included in the pipette solution. Interestingly, this compound failed to eliminate the effect of IP3 to restore the Ca2+-dependent inhibition in OAG-induced ITRPC7 (Figure 6A). The similar results were obtained when TG (2 µmol/L) was present in the bathing solution (Figure 6B). Taken together, these data strongly suggest that the role of IP3R would be, if any, only minor in mediating the effect of IP3.
Discussion
Inactivation/desensitization is the gating process in which channels become non-conductive to intensive and/or prolonged activation stimuli. The biological significance of this process is to restrict the excessive ion flux and maintain the pertinence of chemical and electrical signals. A multitude of factors are known to participate in the inactivation process, such as membrane potential, ions, calmodulin, lipids, and kinases. Amongst these, the Ca2+-dependent inactivation is the common feature observed for many Ca2+-permeable cation channels, including L-type Ca2+ channels, the NMDA receptor, and TRP channels, where PKC often plays a pivotal role[7,21–23]. The TRPC7 channel is subject to the negative regulation of PKC according to Shi et al[15], where their conclusion was made by comparison of the current density of ITRPC7 with either the activator or inhibitor of PKC in the pipette solution that contains different concentrations of Ca2+ (Figure 9C, right column), but the source of Ca2+ (Ca2+ entry or Ca2+ release) that activates PKC remains elusive. One notable finding of the present study is that Ca2+ entry through the activated TRPC7 channel can activate PKC even during a brief application of a physiological concentration of Ca2+o after the full activation of the channel (Figure 1).
There is a common CaM/IP3 receptor (IP3R) binding domain on the C terminus of all TRPC subfamily members[24]. It is proposed that IP3R and CaM competitively bind to this domain, thereby regulating the channel activity[25]. According to Shi et al[15], Ca2+/CaM is a prerequisite for the activation of TRPC6 channels, whereas it exerts tonic inhibition on TRPC7 channels, as evidenced by the increased macroscopic TRPC7 current (by approximately 20%), partial relief from Ca2+o-induced inhibition, and the rightward shift of Ca2+ sensitivity of single TRPC7 channels by treatment with calmidazolium (a specific CaM inhibitor) or co-expression of Ca2+-insensitive mutant CaM. However, the sustained inhibition of ITRPC7 presented after the washout of Ca2+o, which was not examined in detail in this study, could not be relieved by inhibiting CaM. As clearly demonstrated in Figure 1F, the co-expression of mutCaM failed to recover the TRPC7 channels from Ca2+o-induced inhibition, which was instead reverted by an organic PKC inhibitor calphostin C, the PKC-specific inhibitory peptide, or vigorous intracellular Ca2+ buffering (Figures 1,2). These results strongly suggest that Ca2+ entering through the activated TRPC7 channel during receptor stimulation can give rise to 2 different modes of the channel inactivation, one being rapid and dependent on CaM, while the other persisting via the activation of PKC.
With respect to the mechanism responsible for the PKC-mediated inhibition of the TRPC7 channel, a noteworthy finding is the differential reversibility from the Ca2+o-induced inhibition of CCh and OAG-induced currents (Figures 1,2,3,4). Despite both agonists being capable of evoking ITRPC7, recovery from the Ca2+o-induced inhibition was much faster and much more complete with OAG than CCh. However, after the intracellular perfusion of IP3, the recovery of OAG-induced ITRPC7 from the inhibition became as comparably slow as CCh used to evoke the current (Figure 5). The action of IP3 seemed to be specific and direct since its effect was not mimicked by its 2 main metabolites, IP4 and IP2. Furthermore, this effect of IP3 was not prevented by heparin or TG (Figure 6). Considering that both OAG and CCh can activate PKC, the main difference must reside on the actions of IP3, which is only produced in response to CCh stimulation. Therefore, one plausible picture that we envisage is that Ca2+ entry through the activated TRPC7 channel facilitates the activation of PKC with DAG, and consequent phosphorylation of the channels by the activated PKC leads to their inactivation. IP3 may be necessary to stabilize this “inactivation” state, for example, by inhibiting the dephosphorylation of the channel protein via phosphatases, thereby causing a retarded recovery from the inactivation. Importantly, this stabilization seems to occur through an as yet unknown IP3R-independent mechanism. A similar but oppositely-operating synergism between DAG and IP3 has been observed for the activation process of the recombinant TRPC7 channel[15] and a native TRPC6-like channel, α1-adrenoceptor cation channel[26], where IP3 potentiates the extent of channel activation evoked by DAG in a heparin-insensitive manner[26]. However, IP3 has also been reported to activate the TRPC7 channel in an IP3R-dependent manner in chicken DT40 cells, when the channel is expressed at a low level[27]. Such complex aspects of the TRPC7 channel activation and inactivation make it difficult to examine the mechanisms underlying it separately. In addition, since the effect of IP3 on targets other than IP3R is just recently emerging[28], there is no evidence yet available for exploring the molecular basis for the extra-IP3R actions of IP3 on the TRPC7 channels. It would thus be better in future to determine whether these putative IP3-dependent regulatory sites exist on TRPC channels per se, auxiliary proteins, or both, and then to elucidate their molecular identifications. It may also be worthwhile to explore the mechanism underlying Ca2+-induced inhibition observed in this study, by means of mutagenesis of a PKC phosphorylation motif which has commonly been identified on the C-terminus of theTRPC3/6/7 subfamily (Ser712 in TRPC3)[29].
In conclusion, the present study has revealed a novel mechanism for the negative regulation of the TRPC7 channels by intracellular Ca2+ via PKC activation, which requires the cooperative action of DAG and IP3. This mechanism would act as an effective brake against the excessive or prolonged activation of TRPC7 during receptor stimulation, and may be important to regulate a number of pathophysiological processes, such as apoptosis, where the TRPC7 channel likely plays a certain role[30,31]. Further studies, such as a phosphorylation analysis of the activated channel and molecular elucidation of the cooperation of DAG and IP3 by extensive screening of responsible proteins, may help to promote our understanding about the inactivation mechanism of TRPC7.
Acknowledgement
We thank Prof Yasuo MORI (University of Kyoto) for providing the plasmids of TRPC7 and mutCaM.
References
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev 2002;82:429-72.
- Montell C. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci STKE. , 1–17.www.stke.org/cgi/content/full/OC_sigtrans;2001/90/re1
- Clapham DE, Julius D, Montell C, Schultz G. International union of pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels Pharmacol Rev 2005;57:427-50.
- Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of TRP channels. Annu Rev Physiol 2006;68:685-717.
- Lacinova L, Hofmann F. Ca2+- and voltage-dependent inactivation of the expressed L-type Ca(v)1.2 calcium channel. Arch Biochem Biophys 2005;437:42-50.
- Stotz SC, Zamponi GW. Structural determinants of fast inactivation of high voltage activated Ca2+ channels. Trends Neurosci 2001;24:176-81.
- Cens T, Rousset M, Leyris JP, Fesquet P, Charnet P. Voltage- and calcium-dependent inactivation in high voltage-gated Ca2+ channels. Prog Bioph Mol Biol 2006;90:104-17.
- Findlay I. Physiological modulation of inactivation of L-type Ca2+ channels: one switch. J Physiol 2003;554:275-83.
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat activated ion channel in the pain pathway. Nature 1997;389:816-24.
- Szallasi A, Cruz F, Geppetti P. TRPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med 2006;12:545-54.
- Tominaga M, Tominaga T. Structure and function of TRPV1. Pflugers Arch 2005;451:143-50.
- Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M. Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci USA 2003;100:8002-6.
- Docherty RJ, Yeats JC, Bevan S, Boddeke HW. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurons from adult rats. Pflugers Arch 1996;431:828-37.
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 2002;35:721-31.
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, et al. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol 2004;561:415-32.
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Ann Rev Neurosci 2004;27:107-44.
- Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res 1999;33:49-78.
- Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res 2000;87:1095-102.
- Albert AP, Large WA. Signal transduction pathways and gating mechanisms of native TRP-like cation channels in vascular myocytes. J Physiol 2006;570:45-51.
- Hardie RC. TRP channels and lipids: from Drosophila to mammalian physiology. J Physiol 2007;578:9-24.
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron 1998;21:443-53.
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate calcium-dependent inactivation of NMDA receptors. J Neurosci 1999;19:1165-78.
- Nilius B, Voets T. Diversity of TRP channel activation. Novartis Found Symp 2004;258:140-9.
- Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem 2001; 276: 21 303–10.
- Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, et al. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci USA 2001;98:3168-73.
- Albert AP, Large WA. Synergism between inositol phosphates and diacylglycerol on native TRPC6-like channels in rabbit portal vein myocytes. J Physiol 2003;552:789-95.
- Vazquez G, Bird GS, Mori Y, Putney JW Jr. Native TRPC7 channel activation by an inosital trisphosphate receptor-dependent mechanism. J Biol Chem 2006; 282: 25 250–8.
- Kanematsu T, Takeuchi H, Terunuma M, Hirata M. PRIR, a novel Ins(1,4,5)P3 binding protein, functional significance in Ca2+ signaling and extension to neuroscience and beyond. Mol Cells 2005;20:305-14.
- Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW Jr. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol 2005;67:558-63.
- Foller M, Kasinathan RS, Duranton C, Wieder T, Huber SM, Lang F. PGE2-induced apoptotic cell death in K562 human leukaemia cells. Cell Physiol Biochem 2006;17:201-10.
- Satoh S, Tanaka H, Ueda Y, Oyama J, Sugano M, Sumimoto H, et al. Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol Cell Biochem 2006;294:205-15.