
Adenovirus viral interleukin-10 inhibits adhesion molecule expressions induced by hypoxia/reoxygenation in cerebrovascular endothelial cells1
Introduction
Stroke is one of the most frequent causes of death and disability worldwide and has significant clinical and socioeconomic impact. Although different mechanisms are involved in the pathogenesis of stroke, there is increasing evidence showing that inflammation accounts for its progression[1,2]. Inflammation is characterized by the accumulation of inflammatory cells and mediators in the ischemic brain[3]. After the onset of ischemia, inflammatory cells, such as blood-derived leukocytes and microglia are activated and accumulate within the brain tissue, subsequently leading to inflammatory injury. Experimental studies have suggested that polymorphonuclear leukocytes play a role in the development of secondary injury after acute ischemic infarction. The inhibition of the accumulation of neutrophils decreases ischemic injury[4]. Experimental observations have led to the hypothesis that focal cerebral ischemia induces an activated state of leukocytes and cerebral endothelial cells[5]. Subsequently, also on reperfusion, leukocytes may adhere to the activated endothelium, plug capillaries, migrate into brain tissue, and release pro-inflammatory mediators. This inflammatory reaction could lead to secondary injury of potentially salvageable neurons in the penumbra around the infarct[6].
Adhesion molecules play a pivotal role in the infiltration of leukocytes into the brain parenchyma after stroke and may represent important therapeutic targets[7]. Three major steps, rolling, adhesion, and transendothelial migration of leukocytes, are involved in the access of leukocytes to the brain through the endothelial wall. Activated leukocytes, especially neutrophils, result in further damage of ischemic lesions through reperfusion or secondary injury mechanisms[8]. The interaction between leukocytes and the vascular endothelium is mediated by 3 main groups of cell adhesion molecules: selectins, the immunoglobulin superfamily, and integrins[9]. Several reports have shown that inhibiting leukocyte adhesion by targeting various adhesion molecules, thus preventing leukocytes from entering ischemic brain, results in reduced neurological injury[10]. Furthermore, animals deficient in adhesion molecules have a reduced infarct volume after transient focal cerebral ischemia[11]. Several adhesion molecules have been documented in both permanent and transient middle cerebral artery occlusion[3,12]. Animal experiments investigating the effect of anti-adhesion molecule strategies in focal cerebral and spinal cord ischemia have shown a beneficial effect in models in which transient focal ischemia was followed by reperfusion[13,14]. Treatment with anti-adhesion molecule antibodies not only improved outcomes, but also increased the therapeutic time window.
Therefore, the inflammatory response that exacerbates cerebral injury by hypoxia/reoxygenation (H/R) is an attractive therapeutic target. Several studies have confirmed that anti-inflammatory therapies with a deduction of adhesion molecule expressions and leukocyte adhesion rates can significantly attenuate the development of brain infarction and the worsening of the neurological outcomes[15–17].
Our previous studies showed that some drugs could attenuate inflammatory responses and subsequently ameliorate brain ischemia injury by upregulating interleukin-10 (IL-10) expression. It is generally known that IL-10 is a potent anti-inflammatory cytokine produced by Th2 cells that predominantly mediates immunosuppressive effects through the downregulation of macrophage functions and the inhibition of pro-inflammatory cytokines produced by Th1 cells.
Viral interleukin-10 (vIL-10), a known prominent anti-inflammatory cytokine, is highly homologous to human and mouse IL-10 (mIL-10) and shares most functions and many biological properties[18]. Because B lymphocytes play a role in autoimmune diabetes, this unique property of vIL-10 may be especially important[18]. Experimentally, vIL-10 can be distinguished from mIL-10 in mouse models and can be specifically neutralized by an antibody without cross-reactivity to mIL-10. Moreover, another advantage of using vIL-10 is lack of local inflammation at the administration site. These data suggest that the use of vIL-10 instead of mIL-10 provides a more direct and specific experimental assessment in this study.
In this study, we constructed a recombinant adenovirus expressing vIL-10, encoded by the Epstein–Barr virus BCRF1 open-reading frame. The aim was to investigate the effects of the gene transfer of vIL-10 on adhesion molecule expressions and the leukocyte adhesion rates in cerebrovascular endothelial cells injured by H/R.
Materials and methods
Reagents A primary antibody for ICAM-1 and VCAM-1 were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). The mIL-10 ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA). The vIL-10 ELISA kit was obtained from Pharmingen (San Diego, CA, USA).
Construction of a recombinant adenovirus expressing vIL-10 and green fluorescent protein Recombinant adenoviruses were constructed by using a simplified homologous recombination method as previously described[19]. To generate recombinant adenovirus vectors expressing vIL-10, cDNA sequences encoding vIL-10 were isolated from the MFG/vIL10 plasmid. A recombinant adenovirus expressing the green fluorescent protein (GFP) gene or buffer alone was used as a control. High titer adenoviral stocks were propagated in 293 cells and purified by cesium chloride gradient centrifugation. The banded virus was removed, desalted by dialysis in storage buffer (1 mol/L sucrose and 5 mmol/L alpha-cyclodextrin [Sigma, St Louis, MO, USA] in phosphate-buffered saline [PBS]), and stored in small aliquots at -80 °C. Repeated freeze/thaw cycles of the Ad stocks were avoided. Viral stocks and the infected cells were handled only in a class II laminar flow hood and maintained in a CO2 incubator designated for hat purpose. The viral titer was determined by a plaque assay on human embryonic kidney 293 cells. After purification, the virus was suspended in PBS with 3% sucrose and stored at -80 °C until use.
Culture of cerebrovascular endothelial cell line A cerebromicrovascular endothelial cell line from mouse bEnd.3 (American Type Culture Collection, Manassas, VA, USA) was maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% heat inactivated fetal bovine serum, penicillin (0.1 U/L), and streptomycin (0.1 g/L) in a humidified 5% CO2 and 95% air incubator at 37 °C. For adenoviral infections, the cells were grown to 30%–40% confluence and changed to serum-free medium containing Ad/vIL-10 or Ad/GFP. The infected cells were incubated for 1.5 h and then restored to normal DMEM containing 10% fetal calf serum for an additional 2 d of incubation.
H/R After pretreatment for 48 h with Ad/vIL-10, the resulting subconfluent monolayers of bEnd.3 were exposed to hypoxia (5% CO2 and 95% N2) for 9 h in a commercially available hypoxia chamber (model 1029, Forma Scientific, Marietta, Ohio, USA) at 37 °C in the presence or absence of Ad/vIL-10 (multiplicity of infection [MOI]: 10~80) by a grouping progress. After hypoxia incubation, reoxygenation was performed in a humidified cell culture incubator (5% CO2 and 95% air) at 37 °C for 12 h. The culture supernatants were collected for instant analysis or stored at -20 °C for further analysis.
Monocyte adhesion To test the effects of vIL-10 on the adhesion of monocytes to endothelial cells, bEnd.3 cells were grown to confluence on 96-well culture plates and then treated by 9 h hypoxia followed by 12 h reoxygenation before the adhesion assays. Ten minutes before the adhesion assay, the endothelial cells were washed with PBS. Cultured THP-1, a human monocytoid cell line, were washed and diluted to a final concentration of 106 cells/mL. The THP-1 cells (100 μL) were added to each well of the endothelial cells and were allowed to coincubate with the endothelial monolayer for 1 h. Each well was turned to appropriate degrees at 15 min to allow the uniform distribution of the THP-1 cells across the endothelial monolayer. The non-adherent mononuclear cells were carefully removed by 2 washes with preheated PBS. The adherent cells were counted by microscopy using a computer-aided image analysis system (ImageAnalyst, Automatix, Billerica, MA, USA).
ELISA for vIL-10 and mIL-10 The culture supernatants were collected after treatment by H/R. The vIL-10 levels in the supernatants from the fresh endothelial cell cultures were measured using a sandwich ELISA with an anti-vIL-10 monoclonal antibody (mAb; Pharmingen, USA). This antibody did not have cross-reactivity with mIL-10. The ELISA kit for vIL-10 with the mAb was used according to the manufacturer’s directions. Endogenous mIL-10 levels in the culture supernatants were measured using commercially available reagents (R&D Systems, USA).
Western blotting analysis Cultured cells were lysated with 2× SDS (100 mmol/L Tris·HCl [pH 6.8], 200 mmol/L dithiothreitol; 4% SDS, 0.2% bromophenol blue, and 20% glycerin), and centrifuged at 10 000×g for 10 min. The protein concentration was estimated by the BCA protein assay reagent kit (Pierce, Rockford, IL, USA). The samples of 50 µg proteins were then separated by 10% SDS-PAGE with a Bio-Rad mini-protean cell (Hercules, CA, USA), and then transferred to nitrocellulose membranes (Amersham, Arlington Heights, IL, USA). After incubation with blocking solution (5% non-fat milk, Sigma, USA), the membranes were incubated with polyclonal primary antibodies at optional dilution overnight at 4 °C. The membranes were washed and incubated with the second antibody at optional dilution for 2 h or more. They were then detected using an ECL detection system (Amersham Pharmacia Biotech, Uppsala, SE, Sweden), and the relative intensity of the bands of interest was analyzed by relative protein analytic software.
RT-PCR The RNA was extracted with chloroform/isoamyl alcohol and precipitated with 70% ethanol. The resulting RNA was quantified spectophometrically, and then 0.5–1 g/L RNA was used to synthesize the cDNA. The cDNA was amplified using specific primers as follows: vlL-10: forward, 5´-CGAAGGTTAGTGGTCACTCT-3´ and reverse, 5´-CACCTGGCTTTAATTGTCATG-3´; and β-actin: forward, 5´-TAAAGACCTCTATGCCAACACAGT-3´ and reverse, 5´-CTACTCAGGCCGGGGAGGTAGCAC-3´.
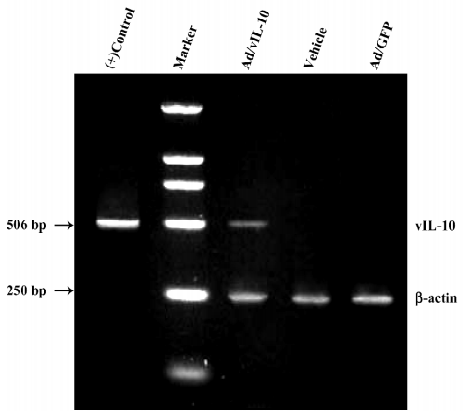
Forty PCR cycles yielded a vIL-10 fragment (506 bp) using an automated thermocycler (PTC100, MJ Research, Hercules, CA, USA) with cycles of 96 °C for 30 s, 58 °C for 45 s, and 72 °C for 1 min. The cDNA from a vIL-10 plasmid using the same RT-PCR reaction was used as a positive control. β-actin (250 bp) was amplified from the same cDNA in a separate reaction and the same PCR cycle settings. The product was separated on a 1% agarose gel and visualized with 1% ethidium bromide staining with a UV source.
Real-time PCR assay ICAM-1 and VCAM-1 mRNA levels were determined by real-time PCR and expressed relative to the expression of GAPDH; the mRNA levels after H/R were normalized to normoxic control bEnd.3. Purified total RNA from bEnd.3 was eluted in RNase-free water and stored at -80 °C. The quality of the samples was determined by analysis on 1.2% agarose electrophoresis gels. Next, total RNA was reverse-transcribed to single-strand cDNA using the SYBR ExScript RT-PCR kit (TaKaRa, Otsu, Japan) by incubation of total RNA (1 µg) with ExScript RTase (100 U), oligo(dT) primer (100 pmol/L), dNTP (1 mmol/L), and a RNase inhibitor (20 U) at 42 °C for 15 min in a final volume of 20 μL. The reaction was terminated by incubation at 95 °C for 2 min. The primers used for the SYBR green PCR are as follows: β-actin: forward, 5´-AGTGTGACGTTGACATCCGT-3´ and reverse, 5´-GCAGCTCAGTAACAGTCCGC-3´; ICAM-1: forward, 5´-GGCATTGTTCTCTAATGTCTCCG-3´ and reverse, 5´-GCTCCAGGTATATCCGAGCTTC-3´; and VCAM-1: forward, 5´-AGTTGGGGATTCGGTTGTTCT-3´ and reverse, 5´-CCCCTCATTCCTTACCACCC-3´.
The ICAM-1 and VCAM-1 primers were obtained from the Primer Bank (http://pga.mgh.harvard.edu/primerbank/). Primers across exon–exon junctions were designed. Contamination with genomic DNA was monitored by omitting the reverse transcriptase, and no-template controls served as the negative control.
SYBR green PCR amplifications were performed in a DNA Engine Peltier Thermal cycler coupled with a Chromo 4 real-time PCR detector (Bio-Rad, USA). The reactions were carried out in a 96-well plate in a 20 µL reaction volume containing 10 µL of 2×SYBR green master mix (TaKaRa SYBR Exscript), a 0.2 µmol/L concentration of each forward and reverse primer. The thermal profile for all SYBR green PCR was 95 °C for 5 min, followed by 40 cycles of 95 °C for 20 s, 60 °C for 40 s, and 72 °C 20 s. Each sample was replicated 2–3 times. All reactions were repeated at least 3 times independently to ensure the reproducibility of the results.
Statistical analysis Data are presented as the mean±SD. Student’s t-test was used for the analysis comparing 2 different groups. Differences between the groups were compared by one-way ANOVA. Statistical significance was considered to be achieved if P<0.05.
Results
Identification of vIL-10 in bEnd.3 after Ad/vIL-10 transfection The RT-PCR analysis revealed that only the vIL-10-transfected group produced a 506 bp fragment consistent with vIL-10 (Figure 1). Neither the vehicle nor GFP controls had any bands when RT–PCR was performed using the specific vIL-10 primers. All of the groups had similar intensity bands of the housekeeping gene β-actin, suggesting adequate starting cDNA.
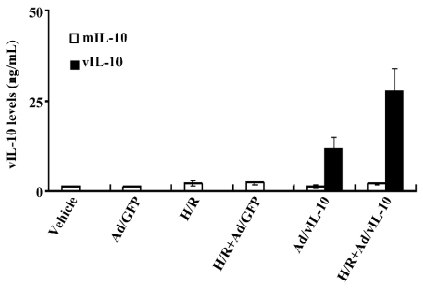
vIL-10 and mIL-10 expressions after Ad/vIL-10 transfection The vIL-10 levels in the supernatant from freshly cultured bEnd.3 were measured using a sandwich ELISA with an anti-vIL-10 mAb (Figure 2). vIL-10 was significantly expressed after the transfection of Ad/vIL-10 (8.62 ng/mL) and further increased by 11.37 ng/mL via H/R treatment. There were no detectable transgene products in the culture medium after the transfection with Ad/GFP. The mIL-10 levels were increased by H/R, but insignificantly, and the gene transfer of vIL-10 or the GFP gene did not significantly affect the mIL-10 levels.
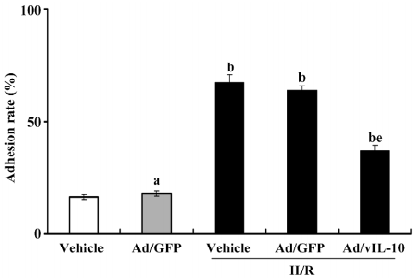
Ad/vIL-10 deducted the adhesion rate of monocytes to cerebrovascular endothelial cells After coincubation for 1 h, the adhesion rate was determined (Figure 3). Ad/GFP transfection did not affect the basic adhesion of monocytes to endothelial cells (P>0.05, 13.26%±1.62% vs 15.33%±1.81% of the vehicle). H/R treatment significantly enhanced the adhesion of monocytes to cultured bEnd.3 (P<0.01, 62.17%±4.73% vs 15.33%±1.81% of the vehicle). There was no significant alteration of the adhesion rate in the Ad/GFP groups treated by H/R as compared with the H/R groups (P>0.05, 58.39%± 5.33% vs 62.17%±4.73%). Ad/vIL-10 significantly inhibited the increase of the adhesion rate by H/R (P<0.05, 31.05%±3.41% vs 62.17%±4.73% of H/R).
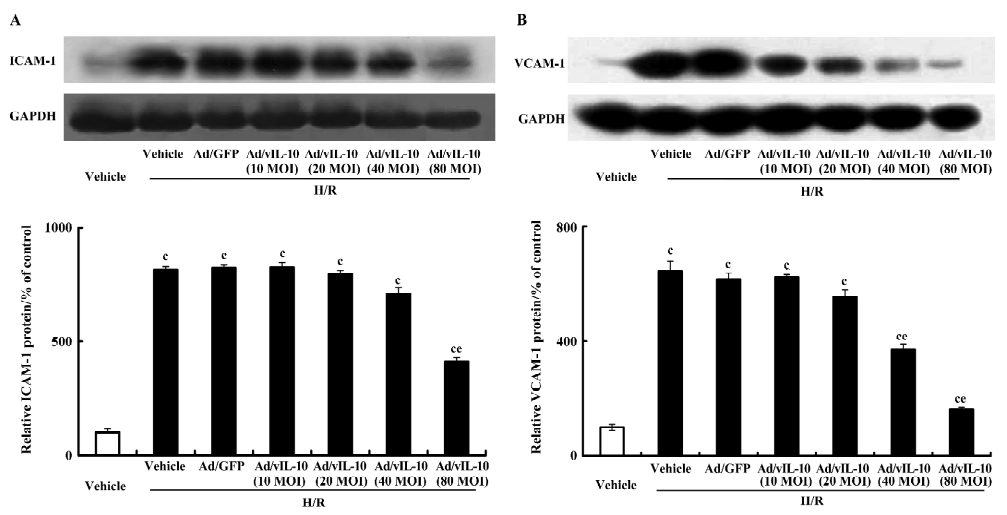
Ad/vIL-10 inhibits adhesion molecule expressions in cerebrovascular endothelial cells Untreated bEnd.3 constitutively expressed ICAM-1 and VCAM-1. After treatment with H/R, the ICAM-1 and VCAM-1 proteins were increased by 8.16- and 6.43-fold, respectively, as compared with the groups not exposed to H/R. Ad/GFP did not affect the expressions of ICAM-1 and VCAM-1 induced by H/R. Ad/vIL-10 (MOI: 10~40) had an insignificant inhibition of the expression of the ICAM-1 protein induced by H/R (P>0.05). After pretreatment with Ad/vIL-10 (MOI: 80), the ICAM-1 protein decreased by 4.13-fold compared with the H/R group (P<0.05; Figure 4A). The VCAM-1 protein level was still higher than that of the vehicle after pretreatment with Ad/vIL-10 (MOI: 40, 80) (3.71- and 1.61-fold vs vehicle, respectively), but was significantly decreased in comparison with that of the H/R-treated groups (P<0.05; Figure 4B).
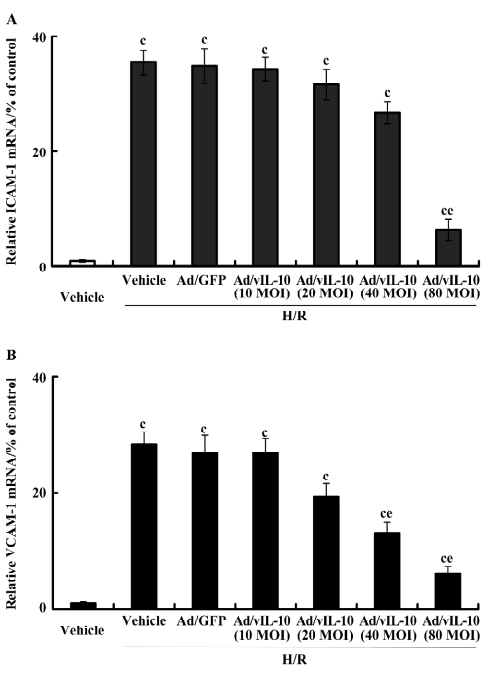
After treatment with H/R, the expressional tendency of ICAM-1 and VCAM-1 mRNA was similar to that of their corresponding protein expressions (Figure 5). Ad/GFP transfection did not affect the basic expression of ICAM-1 and VCAM-1 mRNA. As compared with the vehicle and Ad/GFP groups, the corresponding expressions of ICAM-1 and VCAM-1 were increased by 35.47- and 28.33-fold, respectively, after H/R. H/R did not induce significant alterations of ICAM-1 and VCAM-1 in the Ad/GFP groups in comparison with the H/R-treated groups (P>0.05). ICAM-1 decreased by 6.35-fold by Ad/vIL-10 (MOI: 80, P<0.01; Figure 5A) while Ad/vIL-10 (MOI: 10~40) did not affect ICAM-1 levels after H/R. Ad/vIL-10 (MOI: 20) significantly inhibited VCAM-1 mRNA (P<0.05), while the maximal inhibition of the expression of VCAM-1 was observed at 80 MOI of the Ad/vIL-10 group (P<0.01; Figure 5B).
Discussion
The inflammatory response that exacerbates cerebral injury by H/R is an attractive therapeutic target. The increased expression of adhesion molecules is well known to play an important role in the development of cerebral infarction by the recruitment and transmigration of leukocytes. In this study, we explored the effect of Ad/vIL-10 on monocyte adhesion and the expressions of ICAM-1 and VCAM-1 induced by H/R in cerebrovascular endothelial cells. The results showed that Ad/vIL-10 significantly decreased the adhesion rate of monocytes to cerebrovascular endothelial cells and ICAM-1 and VCAM-1 expressions induced by H/R. These findings indicated that the gene transfer of vIL-10 was an attractive way to inhibit CAM expression, and the inhibition of the upregulation of adhesion molecules was a promising approach to limit inflammatory response during H/R.
In this study, we did not observe any significant increase of mIL-10 in the cellular supernatant by ELISA assay after Ad/vIL-10 or Ad/GFP transfection, which showed that both the viral vector and vIL-10 did not affect the endogenous mIL-10 expression. An insignificant difference in mIL-10 levels was found between the groups treated with H/R and the vehicle, suggesting that the reduction of ICAM-1 and VCAM-1 was specifically produced by vIL-10 and not by endogenous mIL-10. These data showed that the overexpression of vIL-10 independently played a protective role in cell injury induced by H/R under these conditions. The endogenous mIL-10 did not represent a significant protective role and may be related to 2 factors: mIL-10 did not achieve a sufficient concentration and played an inherent role. However, the overexpression of vIL-10 may mask role of mIL-10.
Our data supports the notion that H/R induces endothelial activation and the subsequent upregulation of CAM. After H/R, the adhesion rate increased 46.84%, and the ICAM-1 and VCAM-1 proteins increased by 7.16- and 5.43-fold, respectively, which proved that there was an intrinsic relation between the increase of the adhesion rate and the upregulation of the CAM expression. In the groups pretreated with Ad/vIL-10, there was a significant descent in the adhesion rate and CAM expression. These data showed that direct blocking of CAM or their ligands may represent,, at least in part a potentially effective clinical treatment of ischemia/reperfusion. Moreover, in animal models, the cerebral infarction size was reduced by anti-ICAM-1 antibodies administered during reperfusion[20]. The results from Rentsch et al showed that anti-ICAM-1 antibodies significantly reduced infarction size and number of polymorphonuclear leucocytes in ischemic issues, delayed inflammatory responses during ischemia, and decreased cell apoptosis in the ischemic region[21].
Our data strongly argues that vIL-10 mediates its anti-inflammatory action through the downregulation of CAM expression. In support of this contention, other investigators have found that vIL-10 mediates some of its anti-inflammatory activity through decreased CAM expression[22]. In addition, other models support the interrelation between IL-10 and CAM. IL-10 similarly blunted inflammation secondary to myocardial ischemia/reperfusion through an ICAM-1 mechanism, while in a mouse septic shock model, IL-10 reduced liver injury and mortality through an ICAM-1 and VCAM-1 mechanism. In addition, vIL-10 may not have immunogenicity, and the systemic delivery of replication-deficient adenoviral vectors was well tolerated[18,23]. Therefore, the above-mentioned showed that the gene transfer of vIL-10 was a valuable approach in anti-inflammation and anti-adhesion therapy during brain ischemia/reperfusion injury. We choose gene transfer technology to develop efficient cytokines and prolong the expression of cytokines in vivo because IL-10 has a short circulating half-life, which makes vIL-10 more potent for anti-adhesion and anti-inflammation.
In summary, our results show that Ad/vIL-10 significantly ameliorates monocyte-cerebrovascular endothelial cell adhesion and inhibits the expression of CAM induced by H/R. This study suggests that it is necessary to combine preventing endothelial activation with blocking leukocyte adhesion to avoid inflammation and tissue damage during H/R. Thus, therapy of cerebrovasculature injury during H/R may focus on anti-adhesion and limiting the inflammatory reactions of the organism. These data indicate that the gene transfer of vIL-10 is an attractive approach to ameliorate cerebrovascular endothelium injury during ischemia.
Acknowledgements
We thank Jaculyn DUKE (Dr Paul ROBBINS Laboratory, University of Pittsburgh, Pittsburgh, PA, USA) for providing the plasmids for the reconstruction of adenoviral vIL-10.
References
- Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke 2006;37:291-3.
- Samson Y, Lapergue B, Hosseini H. Inflammation and ischaemic stroke: current status and future perspectives. Rev Neurol (Paris) 2005;161:1177-82.
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003;62:127-36.
- Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet–leukocyte–endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab 2004;24:907-15.
- Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci 2004;9:1339-47.
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol 2007;184:53-68.
- Sughrue ME, Mehra A, Connolly ES Jr, D’Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res 2004;53:497-508.
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal 2001;13:85-94.
- Emsley HC, Tyrrell PJ. Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab 2002;22:1399-419.
- Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke 2004;35:179-84.
- Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res 2004;26:884-92.
- Mrowietz U, Boehncke WH. Leukocyte adhesion: a suitable target for anti-inflammatory drugs. Curr Pharm Des 2006;12:2825-31.
- Relton JK, Sloan KE, Frew EM, Whalley ET, Adams SP, Lobb RR. Inhibition of alpha4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 2001;32:199-205.
- Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke 2001;32:206-11.
- Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol 2001;14:89-94.
- Yang GY, Mao Y, Zhou LF, Ye W, Liu XH, Gong C, et al. Attenuation of temporary focal cerebral ischemic injury in the mouse following transfection with interleukin-1 receptor antagonist. Brain Res Mol Brain Res 1999;72:129-37.
- Zhang L, Zhang ZG, Zhang RL, Lu M, Krams M, Chopp M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 2003;34:1790-5.
- Moore KW, de Waal MR, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001;19:683-765.
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998;95:2509-14.
- Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovasular disease. Stroke 2002;33:2115-22.
- Harlan JM, Winn RK. Leukocyte-endothelial interactions: clinical trials of anti-adhesion therapy. Crit Care Med 2002;30:S214-9.
- Henke PK, DeBrunye LA, Strieter RM, Bromberg JS, Prince M, Kadell AM, et al. Viral IL-10 gene transfer decreases inflammation and cell adhesion molecule expression in a rat model of venous thrombosis. J Immunol 2000;164:2131-41.
- Lindsay JO, Ciesielski CJ, Scheinin T, Hodgson HJ, Brennan FM. The prevention and treatment of murine colitis using gene therapy with adenoviral vectors encoding IL-10. J Immunol 2001;166:7625-33.