
Advances in the genetics of Parkinson's disease1
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder with an overall prevalence of approximately 1.8% of the population over the age of 65 years[1]. PD is characterized by a clinical phenotype consisting of resting tremor, bradykinesia, rigidity, and postural instability, typically asymmetric at onset, gradually progressive, and responsive to dopaminergic therapy[2]. Pathologically, PD is characterized by the loss of dopaminergic neurons in the substantia nigra with intracytoplasmic inclusions containing aggregated α-synuclein as well as other substances (Lewy bodies) and Lewy neurites in many other brain regions as well as in the remaining intact nigral neurons[3]. Motor symptoms are believed to result from the progressive deficiency or dysfunction of dopaminergic neurons in the substantia nigra, regardless of etiology[4].
Relatively little is known regarding the mechanism of PD pathogenesis, in particular the apparent susceptibility of nigral neurons to degeneration. It has been suggested that the selective loss of dopaminergic neurons and the accumulation of α-synuclein is influenced by defects in the ubiquitin-proteasomal system (UPS), mitochondrial dysfunction, and the impairment of mechanisms protecting from oxidative stress and apoptosis[5–7]. These defects in intracellular mechanisms appear to result from a combination of environmental risk factors and genetic susceptibility superimposed on slow, sustained neuronal dysfunction due to advancing ageing.
The contribution of hereditary factors to the etiopathogenesis of PD was proposed well over a century ago, when Leroux[8] and Gowers[9] each noted that a significant percentage of PD patients had an affected family member. A family history of PD is indeed second only to age as a risk factor for the disease[10,11], although familial aggregation of the disease does not necessarily indicate a genetic component, as disease risk may be increased from shared environmental factors. Cross-sectional twin studies reporting similar concordance rates in monozygotic and dizygotic twins challenged the significance of a genetic contribution to PD[12,13], yet suggested that genetic factors may be important when disease begins before the age of 50 years [12]. However, longitudinal twin studies using 18F-dopa positron emission tomography (PET), highlighting clinically presymptomatic dopaminergic loss, reported a concordance rate of 75% in monozygotic twins, compared to 22% in dizygotic pairs, regardless of age at onset[14] supporting a genetic etiology and suggesting that the initial low concordance reported in MZ twins was likely the result of age-related disease penetrance[15].
The past decade has seen a major breakthrough in the genetics of PD, with the identification of 12 genomic regions and pathogenic mutations in 8 genes unequivocally linked to familial PD. The monogenic forms of PD display autosomal dominant and autosomal recessive modes of inheritance, and account for 1%–3% of late-onset disease and approximately 20% of young-onset disease[16,17].
Gene products linked to monogenic forms of PD
Dominantly-inherited PD Dominant forms of familial PD are caused by mutations in SNCA[18,19], UCH-L1[20], and LRRK2[21,22] genes, with probable gain of function effects. Mutations in these genes have also been identified in sporadic late onset PD, suggesting that their protein products might have implications for mechanisms underlying the common idiopathic forms of PD (Table 1).
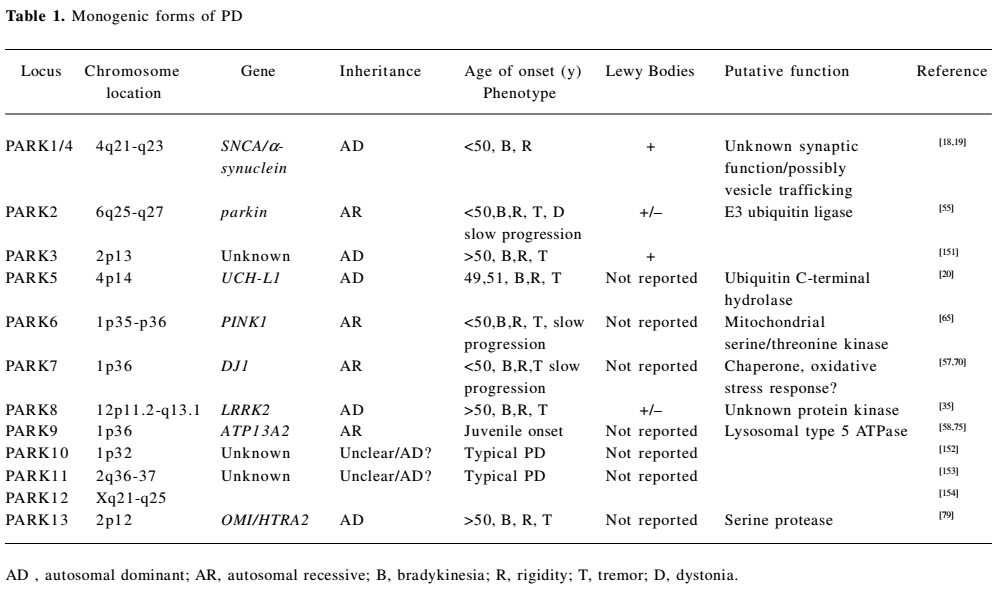
Full table
SNCA (PARK1/4) Mutations in the SNCA gene, encoding α-synuclein, were the first familial PD-associated mutations reported, identified in a large kindred with autosomal dominant PD[18]. Three point mutations that segregate with familial PD have now been identified, Ala53Thr[18], Ala30Pro[23], and Glu46Lys[24].
SNCA gene mutations are rare, accounting for less than 1% of Parkinson disease in the general population[25]. PD patients carrying SNCA mutations have clinically typical PD, with levodopa responsiveness, although disease onset is earlier than in patients with idiopathic PD, and progression appears to be more rapid. Neuropathological findings are similar to those in idiopathic disease, with cell degeneration, Lewy bodies, and neurites. Increased dosage of the wild-type SNCA gene, by either duplication or triplication, has been associated with PD in unrelated families[19,26]. SNCA dosage appears to be correlated with phenotypic severity and age of onset of the disease, with triplication causing rapidly progressive parkinsonism with an early age at onset and early death[19], while SNCA duplication resembles idiopathic PD, with a late age at onset and slower disease progression[26]. Common SNCA gene variants, including a dinucleotide repeat sequence (REP1) within the promoter, have been implicated in increased risk for idiopathic PD[27,28]. Recently, a large meta-analysis confirmed the association between allele-length variability in the dinucleotide repeat and increased PD risk[25].
The normal function of α-synuclein is not fully understood, but evidence suggests it plays a role in vesicular function and may also have chaperone properties[29]. α-Synuclein is a major component of Lewy bodies in both familial and sporadic PD[30]. The three SNCA point mutations alter the properties of the α-synuclein protein, leading to increased protein aggregation, likely critical to Lewy body formation and PD pathogenesis[31].
Ubiquitin carboxy-terminal hydrolase L1 (UCH-L1; PARK5) The ubiquitin–proteasome system has been repeatedly implicated in PD, and the analysis of genes encoding proteins in this pathway in 72 PD families revealed a single missense mutation (Ile93Met) in the ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) gene[20].This mutation was detected in a single sibling pair, the only PD patient carriers of this mutation identified to date. PD in these two patients resembled idiopathic disease, and neuropathology was not available. Subsequent screenings in multiple PD cohorts suggest that a common polymorphism, Ser18Tyr, in the UCH-L1 gene has a protective effect in PD[32]. UCH-L1 is abundant in the human brain and is a component of Lewy bodies[33]. This, together with its involvement in the ubiquitin-proteosomal system[34], supports UCH-L1 as a convincing candidate gene for PD.
LRRK2 (PARK8) PARK8 was originally detected in the large Japanese Sagamihara kindred with autosomal dominant parkinsonism[35], and mutations in leucine-rich repeat kinase 2 (LRRK2) were subsequently identified in a number of families with late-onset autosomal dominant parkinsonism, including the Sagamihara kindred[21,22,36]. Interestingly, mutations in this gene have also been found in late-onset PD patients without a known family history of PD[37,38], suggesting that even late-onset apparently sporadic PD might have a significant genetic component[39].
Mutations in the LRRK2 gene are the most common genetic determinant of PD identified to date[16]. Thus far, at least 20 LRRK2 mutations have been implicated in LRRK2-linked PD estimated to account for approximately 7% of familial PD cases and up to 3% of apparently sporadic disease. While eight mutations which lead to amino acid substitutions seem to be pathogenic, segregating with PD, the others, found in small families or single individuals, are considered putatively pathogenic[40,41]. Gly2019Ser, the most common pathogenic LRRK2 mutation, has been associated with disease at varying frequencies in populations worldwide, and is particularly prevalent among Ashkenazi Jews[42–44] and North African Arabs[45,46]. Recently, a significant association has been reported between the LRRK2 Gly2385Arg missense variant and PD in Asian populations. Originally identified as putative pathogenic mutation in a small Taiwanese PD family[40], LRRK2 Gly2385Arg was subsequently reported as a common polymorphism and not a pathogenic mutation, significantly more frequent among patients with PD than controls in ethnic Chinese populations from Taiwan and Singapore[47–50]. This variant has also been suggested as a risk factor for sporadic PD in the Japanese population[51]. Considering the size of the ageing Asian population, the LRRK2 Gly2385Arg variant is probably the most frequent genetic risk factor for PD worldwide[49,50].
LRRK2-associated PD is clinically indistinguishable from idiopathic disease[52], although substantial variations in neuropathological findings has been reported, including pure nigral degeneration without LB and nigral degeneration associated with brainstem LB typical of PD[22,53].
LRRK2 is a large gene encoding Lrrk2, predicted to contain five functional domains which are believed to be involved in multiple functions, including substrate binding, protein phosphorylation, and protein–protein interactions. All of the identified mutations occur in predicted functional domains[41]. The most frequent pathogenic mutation, Gly2019Ser, occurs in the kinase domain and has been shown to increase kinase activity[54].
Recessively-inherited PD Autosomal recessive (AR) forms of familial PD are caused by homozygous and compound heterozygous mutations in parkin[55], PTEN-induced kinase 1 (PINK1)[56], DJ1[57], and ATP13A2[58]. These relatively rare mutations, with probable loss-of-function mechanisms, are a significant cause of early-onset PD (< age 45 years; Table 1).
Parkin (PARK2) Mutations in the parkin gene were first described in consanguineous Japanese families with autosomal recessive juvenile parkinsonism (AR-JP)[55]. Parkin mutations are the most common cause of AR-JP PD and have been estimated to account for close to 50% of familial patients with recessive inheritance and disease onset before the age of 45 years, as well as 18% of the early-onset apparently sporadic cases[59]. Over 100 mutations in parkin, including exonic deletions, insertions, and point mutations have been observed in patients of all ethnic backgrounds[60]. It has been suggested that parkin alterations may also manifest in an autosomal dominant pattern of disease inheritance, with a single alteration possibly increasing the susceptibility for PD[61–63].
The clinical phenotype of parkin-associated PD varies, although it most commonly resembles idiopathic PD. Features characteristic of parkin disease include early onset, symmetrical motor symptoms, dystonia, improvement of symptoms after sleep, and hyperreflexia with relatively slow progression. Neuropathological studies of patients with parkin mutations with homozygous exonic deletions show selective cell loss of the nigrostriatal tract and locus ceruleus, with a remarkable absence of Lewy bodies, suggesting that parkin disease is a unique entity unlike sporadic PD[16]. α-Synuclein- and ubiquitin-positive inclusions have been reported in patients bearing compound heterozygous deletions and/or mutations[63].
The parkin protein is an ubiquitin E3 ligase preparing target proteins for degradation mediated by the ubiquitin-proteosomal system[64]. Although several putative parkin substrates have been identified, including proteins that are implicated in PD, it is unclear whether there is a single pathological substrate whose accumulation, due to the disrupted enzymatic function of parkin, is responsible for the neuronal death in the substantia nigra[7,60].
PTEN-induced kinase 1 (PINK1; PARK6) The PARK6 locus at 1p35-36 was mapped in a Sicilian kindred segregating an AR form of PD, with a relatively early age of onset[65], and was confirmed in additional European families[66]. Subsequently, mutations in the PTEN-induced kinase 1 (PINK1) gene were identified in three of the PARK6-linked families[56]. Mutations in PINK1 are a rare cause of early-onset PD, most likely accounting for 1%–2% of cases[39]. PINK1 mutations have been identified in families from different European and Asian countries as well as in North American families, indicating that mutations in the gene cause PD in a wide range of populations worldwide[31]. Interestingly, PINK1 mutations have also been found as a rare cause of sporadic early-onset PD[67].
Families with PINK1 disease had a variability in the age of disease onset, with at least one individual in each of the linked families with an age at onset of younger than 45. In family members with late-onset PD, the disease phenotype was identical to that of idiopathic PD, although it appears that the phenotype varies in different populations, with Asian patients experiencing earlier disease onset and more frequent occurrence of atypical symptoms, such as dystonia at onset, hyperreflexia, and psychiatric disturbances[31]. The neuropathology of PINK1 has not been described. Although 18F-dopa PET studies have demonstrated nigrostriatal dopaminergic dysfunction in PINK1 heterozygous carriers[68], as with heterozygous parkin mutations, it is not clear if single PINK1 mutations increase the risk for PD[16].
PINK1 encodes is a highly conserved, widely expressed mitochondrial kinase that has been suggested to protect neurons from stress-induced mitochondrial dysfunction. Mutations in PINK1 increase cell susceptibility to stress conditions, inducing mitochondrial dysfunction and apoptosis[56], thus strengthening hypotheses linking PD to impaired mitochondrial activity and oxidative stress[69].
DJ1 (PARK7) PARK7, a second locus on 1p, was identified in a consanguineous pedigree from the Netherlands[70], and the DJ1 gene was mapped to this locus[57]. Two loss of function DJ1 mutations have been found in PD patients, a deletion of several of exons, preventing DJ1 synthesis, and a point mutation at a highly conserved residue (Leu166Pro) that reduces the stability of DJ1 and promotes degradation through the ubiquitin-proteosomal system, significantly reducing DJ1 levels[71]. DJ1 mutations are rare, accounting for less than 1% of early-onset PD[71].
DJ1 PD is characterized by early onset, asymmetry, and slow progression with a good response to levodopa. The neuropathology associated with DJ1 disease has not been described. Heterozygosity does not likely increase susceptibility for PD, as nearly complete loss of DJ1 protein function is necessary to cause the disease[72].
DJ1 encodes a mitochondrial protein that may be an oxidative stress sensor within cells[73], further supporting a link between mitochondrial impairment and the pathogenesis of PD.
ATP13A2 (PARK9) Kufor–Rakeb syndrome is an AR, juvenile-onset multisystemic neurological disorder whose clinical phenotype includes akinetic-rigid parkinsonism with a good response to levodopa, pyramidal tract dysfunction, supranuclear gaze paresis, and dementia[74]. Kufor-Rakeb was originally detected in a consanguineous Jordanian family[74], was mapped to a 9-cM region of chromosome 1p36[75], and later designated PARK9. Despite the distinct Kufor-Rakeb phenotype, overlap with the disease linked to other PARK loci and the striking response to levodopa in Kufor-Rakeb patients deemed the PARK9 designation appropriate. The causative gene underlying PARK9 was recently revealed to be ATP31A2[58]. Mutations in the ATP13A2 gene were identified in a large non-consanguineous Chilean family and in the original Jordanian kindred[58] and were confirmed in a juvenile-onset PD patient from Brazil[76].
ATP13A2 encodes a large protein belonging to the group 5 P-type ATPase class and displays lysosomal localization in overexpression studies[58]. The function and substrate specificity of this protein are currently undefined, although interestingly, ATP13A2 mRNA is highly expressed in all brain regions, including the substantia nigra, and is upregulated in the human postmortem midbrains from individuals with common idiopathic PD, compared to comparable substantia nigra dopaminergic neurons from controls[58].
Other PARK loci: OMI/HTRA2 (PARK13) Data from animal models have implicated the Omi/Htra2 gene in neurodegeneration with parkinsonian manifestations[77,78]. The OMI/HTRA2 candidate gene was screened in a German PD patient population and in a large ethnically- and gender-matched control sample[79], revealing a novel mutation, Gly399Ser, in 4 PD patients, but in none of the healthy controls. A novel Ala141Ser polymorphism was also associated with PD risk in this population, detected in the heterozygous state in 6.2% of the PD patients compared to 3% of the controls. Heterozygous carriers of both genetic alterations had late-onset PD with typical clinical features and a good response to levodopa therapy.
OMI/HTRA2 encodes the serine protease Omi/HtrA2[80,81]. Interestingly, Omi/HtrA2 has been identified as a component of Lewy bodies in brain samples from pathologically-confirmed PD patients[79]. The Gly399Ser and Ala141Ser mutations result in the defective activation of the proteolytic activity of Omi/HtrA2, compromised mitochondrial function and morphology, and increased vulnerability to cellular stress[79].
Causative genes have not yet been discovered in the dominantly-transmitted PARK3 locus, the PARK12 locus on the X chromosome, or for the PARK10 and PARK11 susceptibility loci, whose modes of transmission remain unclear.
Pathways of PD pathogenesis: insights emerging from the study of monogenic forms of PD
Although familial forms of PD account for fewer than 10% of all PD cases[82], the delineation of monogenic forms of PD in family studies has allowed insights into the mechanisms of neuronal degeneration, processes likely relevant in sporadic disease as well. The molecular functions of the SNCA, parkin, PINK1, DJ1, LRRK2, OMI/HTRA2, and ATP13A2 gene products highlight mitochondrial impairment, oxidative stress, and aberrant protein handling as key events in neuronal dysfunction and degeneration.
Compromise in mitochondrial metabolism, particularly due to defects in complex-I, the first complex of the electron transport chain, was suggested in the pathogenesis of PD long before the identification of disease-causing genes[83]. Postmortem studies have shown mitochondrial impairment and oxidative damage in PD brains[84]. Furthermore, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, and paraquat, noted environmental PD risk factors, are complex-I inhibitors that lead to the aggregation of α-synuclein in vitro and in animal models[85,86]. The aggregation of α-synuclein, downstream to mitochondrial dysfunction, might overwhelm the UPS, allowing the further, possibly toxic, accumulation of proteins that would otherwise be targeted for degradation[87]. PINK1, DJ1, and parkin contribute to mitochondrial protection against oxidative stress, and disease-linked mutations in genes encoding these proteins may compromise mitochondrial integrity, resulting in increased levels of free radicals, as well as a failure of cellular energy and subsequently, impaired UPS function. It has been posited that PINK1 and OMI/HTRA2 might share a common pathway in the mitochondrial response to cellular stress and modulation of apoptosis[88]. UPS activity is also influenced more directly by disease-linked mutations in SNCA, parkin, and DJ1. SNCA mutations result in misfolded, abnormally-aggregated α-synuclein overwhelming the UPS, while mutant parkin exhibits reduced UPS-mediated substrate degradation. DJ1 has been suggested to function as a molecular chaperone or protease, refolding or promoting the degradation of misfolded or aggregated proteins, with mutant DJ1 decreasing the effectiveness of the UPS[87]. Mutant ATP13A2, retained in the endoplasmic reticulum and degraded by the proteasome, may also result in UPS overload, resulting in toxic α-synuclein aggregation[58]. Emerging evidence also supports the involvement of the lysosomal system in PD pathogenesis. The lysosomal degradation pathway, an alternative mechanism for the degradation of proteins, lipids, and damaged organelles, participates in α-synuclein clearance[89–91]. Thus, lysosomal dysfunction, possibly elicited by mutant ATP13A2 in PARK9-linked disease[58] or by the mutant lysosomal enzyme glucocerebrosidase (GBA), which is also implicated in PD risk[92], might result in the accumulation of α-synuclein. Finally, although the cellular functions of the protein kinases LRRK2 and PINK1 are not yet fully understood, they are possibly constituents of a secondary messenger cascade that influences the phosphorylation of proteins that accumulate in end-stage disease[16].
Integration of the multiple, divergent PARK loci protein products linked through mitochondrial impairment, oxidative stress, and protein mishandling to the death of dopaminergic neurons, is still schematic, possibly with interplay at multiple levels. Elucidating the function of each gene product and their interactions remains one of the greatest challenges of PD research.
PD candidate genes
Although there has been great progress in the elucidation of monogenic forms of PD, less is known about genetic alterations underlying the common sporadic form of PD. Multiple biologically-plausible candidate genes have been suggested, based on their roles in the proposed pathways of PD pathogenesis, and case-control association studies have been conducted. Candidate genes studied to date, including mainly genes related to dopamine synthesis, transport, and degradation, detoxification of toxins in dopaminergic neurons, mitochondrial metabolism, and genes encoding essential transcription factors or neurotrophic factors involved in the development of the mesencephalic dopaminergic system, are the basis for several excellent reviews[93–96]. Despite the intense examination of tens of putative candidates, only monoamine oxidase B (MAOB) >188 bp allele showed significant association with sporadic PD in meta-analysis[93], while 6 additional genes (DRD2, ND3, BDNF, SNCA, UCH-L1, and Nurr1), showing significant association with sporadic PD, were replicated in several studies[94]. Very recently, a global genetics consortium, including published and unpublished data from diverse sites worldwide, revealed that allele-length variability in the dinucleotide repeat sequence (REP1) of the SNCA gene promoter is associated with an increased risk for PD[25].
Parkinsonism has been described in patients with Gaucher disease (GD), a recessively-inherited deficiency of the lysosomal enzyme GBA[97]. Clinical observations and neuropathological evidence have implicated mutations in the GBA gene in PD susceptibility, although little is understood regarding the cellular and molecular basis of this association[92]. The identification of Lewy bodies in brain samples from GBA carriers (both PD and GD patients), together with emerging evidence linking heterozygous GBA mutations to diverse synucleopathies[98], suggest that α-synuclein perturbations may be critical to the relationship between GBA mutations and PD risk. Recent data have demonstrated the participation of the lysosomal degradation pathway in α-synuclein metabolism[89–91]. Since the lysosome is the site of GBA metabolism, mutant GBA might elicit lysosomal disturbance or interfere with receptor binding at the lysosomal membrane, impairing α-synuclein degradation and clearance, predisposing GBA mutation carriers to aberrant α-synuclein fibrillization, and possibly α-synuclein-mediated toxicity[92].
Findings from the substantial number of genetic association studies performed to date, claiming or refuting associations between putative PD genes and disease risk, have been compiled and are publicly available on the continuously updated Parkinson’s genetics database, PDGene (http://www.pdgene.org/). Similar to the database that comprehensively catalogs all genetic association studies in the field of Alzheimer’s disease (www.alzgene.org)[99], PDGene provides a powerful tool for deciphering the genetics of PD. Also of interest is the Parkinson’s disease mutation database curated by the Parkinson’s Institute, (www.thepi.org/altruesite/files/parkinson/Mutations/new_page_1.html).
The candidate-gene approach to search for PD susceptibility genes has recently been enhanced by the first high-resolution whole-genome association study of PD[100]. This study highlighted 13 polymorphisms, including SNPs, in the PARK10 and PARK11 loci, as potentially significant in PD susceptibility. A subsequent large-scale analysis of over 12 000 patients failed to replicate the 13 implicated SNPs[101]. An additional genome-wide association study was performed in 267 PD patients and 270 neurologically normal controls[102], with its raw data publicly available online at the Coriell Institute website (ccr.coriell.org/ninds/). Very recently, the first meta-analysis of genome-wide association datasets in PD was performed, suggesting several candidate SNPs for further studies in PD susceptibility[103]. We anticipate that future large-scale genome-wide association studies will fully extend genome coverage in order to reveal additional common PD susceptibility genes.
Interplay of genetics and environment in PD risk
A complex interaction of multiple genetic and environmental risk factors is likely to be involved in the development of PD, manifesting in clinical symptoms once a threshold of neuronal loss is exceeded. While the last decade has focused on the identification of PD-related genes, PD was conventionally thought of as a disorder with an environmental cause. In the 1980s, the discovery of a MPTP-induced acute parkinsonian syndrome in intravenous drug users[104] encouraged the search for exogenous toxins underlying PD and parkinsonism, particularly compounds toxicologically or structurally similar to MPTP, including pesticides, such as rotenone and paraquat. Rotenone and a combined paraquat–maneb exposure induced PD-like pathology and motor signs in rodents[105,106] and a meta-analysis of 19 studies suggested a significant association between human exposure to pesticides and the development of PD[107]. Paraquat, the pesticide most often implicated as a potential neurotoxicant, is the only pesticide for which a dose-dependent relationship has been reported between lifetime cumulative exposure and increased PD risk[108].
Other proposed risks include agricultural employment, rural living, and consumption of well water, long-term exposure to specific metals, high fat/high calorie diet, occupational exposure to viral (or other) respiratory infections, and inflammation in the brain in early life as a consequence of either brain injury or exposure to infectious agents. To date, data regarding these risk factors are equivocal. Cigarette smoking, coffee/caffeine intake, vitamin E consumption, and non-steroidal anti-inflammatory drugs all appear to lower PD risk[109].
Emerging evidence suggests that the interplay between environmental exposures and genetic factors may modify the risk for disease in susceptible individuals. A number of studies have reported an association between environmental toxins and polymorphic genes coding for enzymes involved in the metabolism of foreign chemicals or the transport or metabolism of dopamine in PD risk. Case control studies reported increased PD risk in carriers of CYP2D6[110], GSTP1[111], and SLC6A3/DAT1[112] allelic variants exposed to pesticides, and more recently, solvent exposure in GSTM1 null genotype subjects appeared to increase PD risk[113].
Varying prevalence of PD worldwide: genetic or environmental?
The prevalence of PD and PD-associated mutations differ among ethnic groups and geographical locations. Epidemiological studies have suggested that the prevalence of PD is significantly lower in African-Americans[114] and Asians[115] than in Caucasians, with the lowest disease prevalence in mainland China[96]. The disparity in prevalence may be attributed to environmental differences (climate, industrialization, farming practices, and exposure to environmental toxins), cultural differences (calorie/fat intake and consumption of caffeine and tobacco), and genetic differences (polymorphisms and haplotype structure). Regarding Chinese populations, environmental influences appear significant, with a greater PD prevalence in populations in the industrialized Hong Kong and Taiwan than mainland China[96]. Interestingly, genetic changes in the monoamine oxidase B (MAOB), dopamine transporter (SLC6A3/DAT1), and SNCA candidate genes, which are associated with Parkinson’s disease in Caucasian populations, have not been demonstrated in Chinese populations[116–118].
The Gly2019Ser mutation in LRRK2 is another example of a mutation with ethnic variability. The Gly2019Ser change in LRRK2 exon 41 has been associated with the disease at varying frequencies in Asian, European, and North American populations, and is particularly prevalent among North African Arabs (37% in familial PD patients, 41% in sporadic PD patients, and 1% in controls)[45,46] and in Ashkenazi Jews (29.7%, 26.1% and 26% in familial PD patients, 13.3%, 7.7%, and 10.6% in apparently sporadic PD patients, and 1.3%, 2.0%, and 2.4% in control samples[42–44], respectively). Despite a wide spectrum of ethnic backgrounds, carriers of this mutation share common haplotypes, suggestive of a single-founder effect[38,119,120] or two-founder events[121], with the founder possibly originating in the Middle East[42]. The Gly2019Ser mutation is very rare and does not appear to play a role in the causality of PD in Asian subjects [122–124]. Interestingly, the LRRK2 Gly2385Arg alteration was recently shown to be a common risk factor for PD in ethnic Chinese[47–50] and Japanese[51] populations, and appears to be absent in Caucasians[125], highlighting the contribution of specific genetic factors to disease risk in distinct populations.
Pharmacotherapy, pharmacogenetics, and the search for neuroprotective therapy in PD
Dopaminergic neuron degeneration in PD leads to a profound depletion of dopamine, leading to the cardinal motor symptoms that characterize this disease[4]. Current PD therapy is primarily based on neurotransmitter replacement, using levodopa or dopamine agonists[126]. While virtually all patients enjoy a good response to levodopa, one of the criteria for the diagnosis of PD[127], a minority of patients with pathologically-proven PD experience poor or no response. Regrettably, even in levodopa-responsive patients, therapy becomes less effective over time, as the underlying disease progresses. The improvement of motor function gradually diminishes after 2–7 years of therapy, and a significant proportion of patients develop motor response fluctuations in the forms or “wearing off” or “on/off” phenomenon as well as a peak of dose or end of dose involuntary movements called dyskinesias. In the more advanced stages of the disease, levodopa-induced side-effects can be more disabling than the primary symptoms of the disease itself[128]. Young age at onset, disease duration, duration of levodopa treatment, and female sex have been implicated as risk factors for the development of dyskinesias[129]. Drugs from the dopamine agonist group also elicit undesirable side-effects, including extreme somnolence and neuropsychiatric symptoms[126]. Recently, dopamine agonists have been implicated in a severe “dopamine dysregulation syndrome”, and in particular, impulse control disturbances like pathological gambling, hypersexuality, bulimia with weight gain, and excessive drive for money spending[130,131]. Association studies have suggested that functional polymorphisms in genes encoding drug-metabolizing enzymes, drug receptors, and proteins involved in pathway signaling might be important factors in the large inter-individual variability regarding the efficacy of dopaminergic therapy as well as treatment-induced motor complications[94,129]. Advances in the study of pharmacogenetics, the effect of variation in human genetics on variation in response to pharmacological treatment, will eventually influence the choice of PD therapy. Ultimately, a future challenge will be the replacement of standard therapies with novel individualized therapeutic regimes, with maximum efficacy and minimum toxicity.
Non-motor symptoms occur commonly in PD and may be as debilitating as their motor counterparts[126]. These non-motor phenomena, which may occur years before PD diagnosis and frequently complicate advanced disease, include autonomic dysfunction, sleep disturbances, fatigue, mood disorders, and cognitive dysfunction/dementia. These features likely reflect degeneration of non-dopaminergic neurons and may be unaffected or exacerbated by dopaminergic therapies, emphasizing the necessity for additional PD therapeutic options. As the field of PD has possibly reached the limit of symptomatic therapy, the intensive search for neuroprotective therapy based on the protection or rescue of vulnerable neurons and the arrest of disease progression is critical. An appropriate intervention should address the therapeutically challenging non-motor symptoms of PD while prolonging the period of well-controlled motor symptoms. To date, several candidate PD-neuroprotective agents have been tested in clinical trials, although none have unequivocally demonstrated a disease-modifying effect[132,133].
Hypothesis-generating pathogenetic insights from genetic studies also provide the rationale for additional therapeutic approaches, which can be tested in the laboratory setting and perhaps later in mutation-carrier patients and high-risk presymptomatic individuals. Patients with confirmed hereditary PD will likely be a valuable population for the evaluation of neuroprotective candidates and the arrest of motor and non-motor symptom progression. Asymptomatic high-risk individuals, such as carriers of LRRK2 and other genetic causes of PD, will play a critical role in the longitudinal study of disease progress from preclinical to symptomatic stages. Neuroimaging can be implemented to monitor disease progress and the efficacy of potential neuroprotective candidates in this population, who might also serve as a valuable source for the identification and assessment of additional clinically meaningful biomarkers and risk factors. Ultimately, neuroprotection will aim to capture high-risk individuals in the presymptomatic period, exploiting the valuable therapeutic window of about five years from the onset of neuronal loss to the appearance of clinical signs[134].
If the pathogenic mechanisms in monogenic forms of PD are similar to sporadic disease, therapeutics delineated from the study of mutation carriers might also have relevance for the greater sporadic PD population. We have learned from the intense study of human cancer, which in the last three decades have seen the thorough analysis of hundreds of cancer-related genes, that there is a tremendous gap between molecular discoveries and the establishment and approval of novel effective therapy. On the other hand, molecular studies and insight into cellular and molecular pathways underlying the spectrum of human tumorigenesis have generated multiple effective anticancer treatments, including Trastuzumab (Herceptin) in breast cancer[135] and Imatinib (Glivec) in chronic myelogenous leukemia[136]. Hopefully, the identification, establishment, and approval of PD-neuroprotective therapy will be much shorter.
Two putative PD treatment strategies might target the products of the autosomal dominant SNCA and LRRK2 genes. α-synuclein is likely the most promising target for sporadic PD due to its deposition into Lewy bodies. α-synuclein aggregation is believed to be critical for neuronal toxicity, and an effective therapy might lower α-synuclein levels. The controversy regarding which forms of aggregated α-synuclein mediate toxicity, soluble oligomers, or highly insoluble fibrils must first be resolved. Transgenic mice deficient for α-synuclein or expressing mutant (Ala30Pro, Ala53Thr, or both) or wild-type α-synuclein have been generated. Interestingly, while α-synuclein knockouts did not demonstrate any detectable abnormalities other than an alteration of dopamine release in response to rapid stimulation[137], they showed resistance to the dopaminergic toxin MPTP[138], implicating α-synuclein in the pathogenic mechanism that leads to MPTP-induced parkinsonism. While disappointingly, the overexpression models did not display dopaminergic neuronal death in the substantia nigra, they recapitulated a spectrum of neuropathological changes and α-synuclein abnormalities common to human PD[139,140], and are thus relevant for the testing of novel neuroprotective therapies. Studies in mouse and Drosophila models and cultured cells overexpressing wild-type or mutant α-synuclein have revealed a role for the endogenous molecular chaperone heat shock protein 70 (Hsp70) in the protection of dopamine neurons from the cytotoxic effects of α-synuclein overexpression. The naturally-occurring benzoquinone ansamycin, geldanamycin, has been shown to inhibit α-synuclein aggregation and toxicity in Drosophila and in cultured cells, possibly by modulating Hsp70 molecular chaperone activity, and warrants exploration as a neuroprotective strategy in PD[141]. Emerging evidence implicating the malfunction of the lysosomal degradation pathway in α-synuclein aggregation suggests that enhancing lysosomal function might also prove to be an effective neuroprotective intervention[90]. The autophagy inducer rapamycin demonstrated increased clearance of all forms of α-synuclein[89], although its long-term use is associated with many complications. However, novel modulators of autophagy might have therapeutic potential for the treatment of PD. An additional development based on the pathological involvement of α-synuclein aggregation in PD is the development of a PD vaccine. Recently, a vaccine based on human α-synuclein elicited the generation of anti-α-synuclein antibodies in a mouse model[142]. This putative intervention offers hope to high-risk PD-mutation carriers with a positive family history of PD.
Mutations in LRRK2 are the most common genetic determinant of PD identified to date occurring in familial and apparently sporadic PD. Pathogenic LRRK2 mutations alter its kinase activity increased in the case of Gly2019Ser[54] and Ile2020Thr[143], which is in line with an expected gain-of-function mechanism for their dominant transmission. The increased kinase activity of mutant Lrrk2 is postulated to mediate its toxic effects in cell culture, including inclusion body formation and the triggering cell death in neurons[144]. Furthermore, kinase-dead versions of Lrrk2 are reportedly less toxic than their active equivalents, even when pathogenic mutations are present in the molecule outside of the kinase domain[145], suggesting that kinase inhibitors might offer a new therapeutic approach to treat or delay PD progression in patients with LRRK2 mutations and perhaps in sporadic PD as well. The importance of kinase activity in disease pathogenesis remains to be tested in vivo in transgenic animal models with LRRK2 mutations. Current limitations include lack of knowledge regarding substrates for Lrrk2 kinase activity and the deficiency of an available animal model.
Products of the recessive genes might also advance the development of PD therapies. The loss-of-function found in parkin-, DJ1-, and PINK1-linked PD suggests that increased expression of these proteins may prevent or ameliorate the disease in high-risk individuals and delay PD progression in carrier patients[16]. Parkin and DJ1 knockout mice have been developed and might serve as important tools for the study of potential therapeutic options[146]. However, the relative rarity of parkin, DJ1, and PINK1-linked disease, together with their unique phenotypes, challenge their relevance to the development of treatment for sporadic PD[145].
Genetic counseling
With the identification of genes that are implicated in the causation of familial PD, the demand for genetic testing by patients and their family members as well as by physicians will surely increase. Genetic testing refers to the evaluation of an individual’s genetic material (DNA) to determine the predisposition to a specific disease or to confirm the diagnosis of genetic disease. Testing of mutations in known disease-causing genes has been useful in the definition and classification of many heterogeneous inherited neurodegenerative disorders[147]. To date, molecular genetic testing is clinically available for parkin, PINK1[148], and LRRK2 G2019S and I2020T mutations (http://www.genetests.org), although no formal guidelines have been established by any of the international PD alliances. Whole PD gene sequencing allows the identification of alterations in the entire gene, while testing for specific known disease-causing mutations can be performed using multiple assays, such as sequence, TaqMan and others.
Mutation status is particularly important when results have diagnostic or therapeutic consequences. In the case of “suspected PD” in individuals with a questionable extrapyramidal clinical picture, the detection of a disease-causing gene may verify a clinical diagnosis of PD. Unfortunately, testing will not necessarily prove informative, as the failure to detect a mutation does not negate the diagnosis of PD nor does it rule out the presence of a mutation in another region, while conversely, a detected mutation might not prove pathogenic. Furthermore, a better understanding of mutation frequency, age-dependent penetrance, and natural history are necessary in order to better interpret test results and provide effective genetic counseling. Presently, in an era when PD treatment is purely symptomatic, the clinical utility of genetic testing is questionable, as mutation status will not alter the treatment of monogenic forms of PD. The decision to pursue genetic testing should be made on an individual basis and should be accompanied by genetic counseling.
While the advantages of testing asymptomatic family members include close follow up and early detection of PD as well as the opportunity to plan both psychologically and financially for the future, these must be weighed against the negative emotional and social consequences, possible employment and insurance discrimination, and inconclusive counseling due to the partial penetrance and the inadequate understanding of the age-dependant risk of the disease causing mutations. As long as there is no proven medical or behavioral treatment that can modify the natural history of PD, the justification for the clinical testing of asymptomatic family members currently remains highly questionable. The implications of genetic testing in PD are highlighted in comprehensive reviews by McInerney-Leo et al[149], Tan and Jankovic[147], and Klein[150-154].
Conclusions
In the last decade, major advances have been made in understanding the genetic basis of PD. The identification of pathogenic mutations in PARK-linked genes contests the once held environmental hypothesis for PD and brings the scientific community closer to the elucidation of the enigmatic pathogenesis of this common and devastating disorder. Most importantly, understanding the molecular and cellular pathways involved will be critical for the development of desperately needed preventative, symptomatic, and curative treatment modalities.
References
- de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S21-3.
- Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 2003;991:1-14.
- Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 1996;55:259-72.
- Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson’s disease. Hum Mol Genet 1997;6:1687-91.
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003;39:889-909.
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003;302:819-22.
- Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem 2005;74:29-52.
- Leroux PD. . in 1880: Paris.
- Gowers WR. A Manual of Diseases of the Nervous System. Vol 1. Philadelphia:Blakiston’s Son; 1900.
- Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med 1998;339:1044-53.
- Rocca WA, McDonnell SK, Strain KJ, Bower JH, Ahlskog JE, Elbaz A, et al. Familial aggregation of Parkinson’s disease: The Mayo Clinic family study. Ann Neurol 2004;56:495-502.
- Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, et al. Parkinson disease in twins: an etiologic study. JAMA 1999;281:341-6.
- Wirdefeldt K, Gatz M, Schalling M, Pedersen NL. No evidence for heritability of Parkinson disease in Swedish twins. Neurology 2004;63:305-11.
- Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ. The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol 1999;45:577-82.
- Maher NE, Currie LJ, Lazzarini AM, Wilk JB, Taylor CA, Saint-Hilaire MH, et al. Segregation analysis of Parkinson disease revealing evidence for a major causative gene. Am J Med Genet 2002;109:191-7.
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006;7:306-18.
- Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat 2007;28:641-53.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045-7.
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841.
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998;395:451-2.
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44:595-600.
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601-7.
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998;18:106-8.
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164-73.
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 2006;296:661-70.
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004;364:1169-71.
- Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, et al. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet 2001;10:1847-51.
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, et al. alpha-Synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol 2004;56:591-5.
- Lorincz MT. Clinical implications of Parkinson’s disease genetics. Semin Neurol 2006;26:492-8.
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839-40.
- Gosal D, Ross OA, Toft M. Parkinson’s disease: the genetics of a heterogeneous disorder. Eur J Neurol 2006;13:616-27.
- Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, et al. UCHL1 is a Parkinson’s disease susceptibility gene. Ann Neurol 2004;55:512-21.
- Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J Pathol 1990;161:153-60.
- Pickart CM. Ubiquitin in chains. Trends Biochem Sci 2000;25:544-8.
- Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 2002;51:296-301.
- Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 2005;57:918-21.
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005;365:415-6.
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005;76:672-80.
- Gasser T. Genetics of Parkinson’s disease. Curr Opin Neurol 2005;18:363-9.
- Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, et al. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 2005;6:171-7.
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci 2006;29:286-93.
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2006;354:424-5.
- Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 2006;67:1786-91.
- Orr-Urtreger A, Shifrin C, Rozovski U, Rosner S, Bercovich D, Gurevich T, et al. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson’s disease: is there a gender effect? Neurology 2007;69:1595-602.
- Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, et al. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol 2005;58:784-7.
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 2006;354:422-3.
- Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, et al. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 2006;7:133-8.
- Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, et al. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet 2007;120:857-63.
- Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR. A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol 2006;6:47.
- Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord 2007;13:89-92.
- Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, et al. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 2007;18:273-5.
- Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol 2005;57:762-5.
- Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 2004;62:1619-22.
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 2005;102:16842-7.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605-8.
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004;304:1158-60.
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299:256-9.
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006;38:1184-91.
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000;342:1560-7.
- Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet 2004;13:R127-33.
- Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 2000;48:65-71.
- Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 2001;50:293-300.
- Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 2005;58:411-22.
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000;25:302-5.
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 2001;68:895-900.
- Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MM, et al. PARK6-linked parkinsonism occurs in several European families. Ann Neurol 2002;51:14-8.
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, et al. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 2004;56:336-41.
- Khan NL, Valente EM, Bentivoglio AR, Wood NW, Albanese A, Brooks DJ, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol 2002;52:849-53.
- Shen J, Cookson MR. Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron 2004;43:301-4.
- van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet 2001;69:629-34.
- Lockhart PJ, Lincoln S, Hulihan M, Kachergus J, Wilkes K, Bisceglio G, et al. DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J Med Genet 2004;41:e22.
- Dekker MC, Eshuis SA, Maguire RP, Veenma-van der Duijn L, Pruim J, Snijders PJ, et al. PET neuroimaging and mutations in the DJ-1 gene. J Neural Transm 2004;111:1575-81.
- Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA 2004;101:9103-8.
- Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 1994;89:347-52.
- Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wriekat AL, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet 2001;38:680-2.
- Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007;68:1557-62.
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003;425:721-7.
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 2004;24:9848-62.
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet 2005;14:2099-111.
- Faccio L, Fusco C, Chen A, Martinotti S, Bonventre JV, Zervos AS. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J Biol Chem 2000;275:2581-8.
- Gray CW, Ward RV, Karran E, Turconi S, Rowles A, Viglienghi D, et al. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur J Biochem 2000;267:5699-710.
- Dawson TM, Dawson VL. Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest 2003;111:145-51.
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989;1:1269.
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci 2003;991:120-31.
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 2002;277:1641-4.
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci 2003;23:10756-64.
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 2005;28:57-87.
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 2006;7:207-19.
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003;278:25009-13.
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004;24:1888-96.
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004;305:1292-5.
- Hruska KS, Goker-Alpan O, Sidransky E. Gaucher disease and the synucleinopathies. J Biomed Biotechnol 2006; 2006: 78549.
- Tan EK, Khajavi M, Thornby JI, Nagamitsu S, Jankovic J, Ashizawa T. Variability and validity of polymorphism association studies in Parkinson’s disease. Neurology 2000;55:533-8.
- Gilgun-Sherki Y, Djaldetti R, Melamed E, Offen D. Polymorphism in candidate genes: implications for the risk and treatment of idiopathic Parkinson’s disease. Pharmacogenomics J 2004;4:291-306.
- Gandhi S, Abou-Sleiman PM, Healy DG, Weale M, Gilks W, Ahmadi K, et al. Population genetic approaches to neurological disease: Parkinson’s disease as an example. Philos Trans R Soc Lond B Biol Sci 2005;360:1573-8.
- Benmoyal-Segal L, Soreq H. Gene-environment interactions in sporadic Parkinson’s disease. J Neurochem 2006;97:1740-55.
- Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996;89:691-4.
- Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VM, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006;67:908-10.
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007;39:17-23.
- Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, et al. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet 2005;77:685-93.
- Elbaz A, Nelson LM, Payami H, Ioannidis JP, Fiske BK, Annesi G, et al. Lack of replication of thirteen single-nucleotide polymorphisms implicated in Parkinson’s disease: a large-scale international study. Lancet Neurol 2006;5:917-23.
- Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2006;5:911-6.
- Evangelou E, Maraganore DM, Ioannidis JP. Meta-analysis in genome-wide association datasets: strategies and application in Parkinson disease. PLoS ONE 2007;2:e196.
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983;219:979-80.
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000;3:1301-6.
- Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson’s disease? Brain Res 2000;873:225-34.
- Priyadarshi A, Khuder SA, Schaub EA, Shrivastava S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology 2000;21:435-40.
- Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, et al. Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology 1997;48:1583-8.
- Lai BC, Marion SA, Teschke K, Tsui JK. Occupational and environmental risk factors for Parkinson’s disease. Parkinsonism Relat Disord 2002;8:297-309.
- Elbaz A, Levecque C, Clavel J, Vidal JS, Richard F, Amouyel P, et al. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann Neurol 2004;55:430-4.
- Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet 1998;352:1344-6.
- Kelada SN, Checkoway H, Kardia SL, Carlson CS, Costa-Mallen P, Eaton DL, et al. 5' and 3' region variability in the dopamine transporter gene (SLC6A3), pesticide exposure and Parkinson’s disease risk: a hypothesis-generating study. Hum Mol Genet 2006;15:3055-62.
- Dick F, De Palma G, Ahmadi A, Osborne A, Scott NW, Prescott GJ, et al. Gene-environment interactions in parkinsonism and Parkinson’s disease: the Geoparkinson study. Occup Environ Med 2007.
- Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 2003;157:1015-22.
- Zhang ZX, Roman GC. Worldwide occurrence of Parkinson’s disease: an updated review. Neuroepidemiology 1993;12:195-208.
- Mellick GD, Buchanan DD, Silburn PA, Chan DK, Le Couteur DG, Law LK, et al. The monoamine oxidase B gene GT repeat polymorphism and Parkinson’s disease in a Chinese population. J Neurol 2000;247:52-5.
- Leighton PW, Le Couteur DG, Pang CC, McCann SJ, Chan D, Law LK, et al. The dopamine transporter gene and Parkinson’s disease in a Chinese population. Neurology 1997;49:1577-9.
- Chan DK, Mellick G, Cai H, Wang XL, Ng PW, Pang CP, et al. The alpha-synuclein gene and Parkinson disease in a Chinese population. Arch Neurol 2000;57:501-3.
- Lesage S, Leutenegger AL, Ibanez P, Janin S, Lohmann E, Durr A, et al. LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 2005;77:330-2.
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet 2005;42:e65.
- Zabetian CP, Hutter CM, Yearout D, Lopez AN, Factor SA, Griffith A, et al. LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet 2006;79:752-8.
- Lu CS, Simons EJ, Wu-Chou YH, Fonzo AD, Chang HC, Chen RS, et al. The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson’s disease. Parkinsonism Relat Disord 2005;11:521-2.
- Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E, et al. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett 2005;384:327-9.
- Fung HC, Chen CM, Hardy J, Hernandez D, Singleton A, Wu YR. Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson’s disease. Mov Disord 2006;21:880-1.
- Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohe CF, et al. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur J Hum Genet 2006;14:322-31.
- Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson’s disease (2001): treatment guidelines. Neurology 2001;56:S1-S88.
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181-4.
- Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord 2004;19:997-1005.
- Linazasoro G. New ideas on the origin of L-dopa-induced dyskinesias: age, genes and neural plasticity. Trends Pharmacol Sci 2005;26:391-7.
- Weintraub D, Siderowf AD, Potenza MN, Goveas J, Morales KH, Duda JE, et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol 2006;63:969-73.
- Giladi N, Weitzman N, Schreiber S, Shabtai H, Peretz C. New-Onset Heightened Interest or Drive for Gambling, Eating, Shopping or Sexual Activity in Patients with Parkinson's Disease: The Role of Dopamine Agonist Treatment and Age at Motor Symptom Onset. J Psychopharmacology 2007;21:501-6.
- Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, et al. Neuroprotective agents for clinical trials in Parkinson’s disease: a systematic assessment. Neurology 2003;60:1234-40.
- Olanow CW. The scientific basis for the current treatment of Parkinson’s disease. Annu Rev Med 2004;55:41-60.
- Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114:2283-301.
- Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med 2007;357:39-51.
- Hu J, Zhou GB, Wang ZY, Chen SJ, Chen Z. Mutant transcription factors and tyrosine kinases as therapeutic targets for leukemias: from acute promyelocytic leukemia to chronic myeloid leukemia and beyond. Adv Cancer Res 2007;98:191-220.
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239-52.
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci USA 2002;99:14524-9.
- Fernagut PO, Chesselet MF. Alpha-synuclein and transgenic mouse models. Neurobiol Dis 2004;17:123-30.
- Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ. Parkinson’s disease: a rethink of rodent models. Exp Brain Res 2006;173:196-204.
- McNaught KS, Olanow CW. Protein aggregation in the pathogenesis of familial and sporadic Parkinson’s disease. Neurobiol Aging 2006;27:530-45.
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005;46:857-68.
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 2006;15:223-32.
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006;23:329-41.
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol 2006;60:389-98.
- Fleming SM, Fernagut PO, Chesselet MF. Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx 2005;2:495-503.
- Tan EK, Jankovic J. Genetic testing in Parkinson disease: promises and pitfalls. Arch Neurol 2006;63:1232-7.
- Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol 2006;2:136-46.
- McInerney-Leo A, Hadley DW, Gwinn-Hardy K, Hardy J. Genetic testing in Parkinson’s disease. Mov Disord 2005;20:1-10.
- Klein C. Implications of genetics on the diagnosis and care of patients with Parkinson disease. Arch Neurol 2006;63:328-34.
- Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet 1998;18:262-5.
- Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol 2002;52:549-55.
- Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, et al. Significant linkage of Parkinson disease to chromosome 2q36-37. Am J Hum Genet 2003;72:1053-7.
- Pankratz N, Nichols WC, Uniacke SK, Halter C, Murrell J, Rudolph A, et al. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet 2003;12:2599-608.