
Molecular markers and targets for colorectal cancer prevention
Introduction
Cancer is a group of diseases characterized by uncontrolled growth and spread of abnormal cells. It is caused by both external and internal factors. These causal factors may act together or in sequence to initiate/promote cancer. In spite of knowing more than ever about the genetic and cellular events that accelerate or inhibit cancer induction, cancer is still the number one health concern of western countries. Colon cancer is the third most common disease in the United States, prevalent in both men and women. Globally, colon cancer is the fourth most common cancer in men and the third most common cancer in women. In the USA, as per the statistics of the National Cancer Institute of 2007, there are 112 340 new cases of colon cancer, 41 420 new cases of rectal cancer and 52 180 deaths from both cancers combined. In the United States, average-risk patients account for approximately 75% of all colorectal cancers and include persons older than 50 years with no other known risk factor; moderate-risk patients account for 15%–20% of all colorectal cancers and include those with a positive family history of colorectal adenomatous polyps or cancer; and high-risk patients account for 5%–15% of all colorectal cancers and include those with familial adenomatous polyposis (FAP), hereditary nonpolyposis colorectal cancer or long-standing inflammatory bowel disease[1]. The majority of colorectal cancers are thus nonhereditary and sporadic, which makes early detection important. Most of the cases are identified at advanced stages, rendering curative treatment impossible. Hence, attention has most focused on screening for targets to aim through chemoprevention to reduce the number of colorectal cases.
Chemoprevention strategies can be benefited from observations linking the identification of intermediate/surrogate biomarkers or various nutrients/drugs with specific cancer related molecular targets. In the last few years, genetic information about cancer, molecular signaling and metabolic pathways has been translated into therapies that target specific molecules for prevention. Developing new technologies to provide knowledge of SiRNA’s, microRNA’s and piRNAs functions can form a basis for the development of new chemopreventive agents, which can intervene at any time during the process of multistage carcinogenesis to: prevent the initial mutation; block promotion to premalignant tumors; stop progression from the premalignant state to in situ carcinomas; or prevent invasion or metastasis. As the early stages of tumor promotion and progression are rate limiting, successful targeting of molecular events during these stages can have high impact on outcomes.
Molecular biomarkers in colon cancer
Recent advances in cancer pathogenesis help us in unraveling the vital/valid molecular biomarkers involved in colon carcinogenesis. Identification/discovery of molecular biomarkers ranges from exposure assessment, risk assessment and management to clinical trials. This can help us in developing/discovering novel therapeutic interventions, preventive strategies and agents. Along with these, there is also a need to develop and validate molecular biomarkers reflective of exposure and risk from etiological factors and to use these biomarkers for the design and implementation of prevention strategies in community settings. There are sub-sets of high-risk people within populations who are most susceptible to disease. Thus, methods that can identify these high-risk individuals with high specificity and selectivity will greatly facilitate the implementation of a spectrum of targeted prevention techniques directed towards reducing individual risk. Technologies genomics, proteomics and recent developments in miRNAs are guiding us through the development of agents which are able to target the biomarkers. A link between miRNAs function and cancer pathogenesis is supported by studies examining the expression of miRNAs in clinical samples. miRNAs are emerging as a new class of genes involved in cancer. Knowledge about micro RNA’s and its specific functions offers opportunities to target genetic or epigenetic changes that influence cancer risk. As of now, very little information is known about miRNA’s, a more detailed understanding of micro RNA functions is required to identify and manipulate the molecular targets for cancer prevention. Specific molecular processes have been targeted for therapeutic intervention, including growth factor receptors, proliferation signaling, cell cycling, apoptosis, angiogenesis, the immune system, etc. Many cancers are characterized by alternations in certain signaling pathways, and identification of the aberrant pathway in a particular patient allows for targeted therapy to that specific pathway.
Surrogate end point in colon cancer
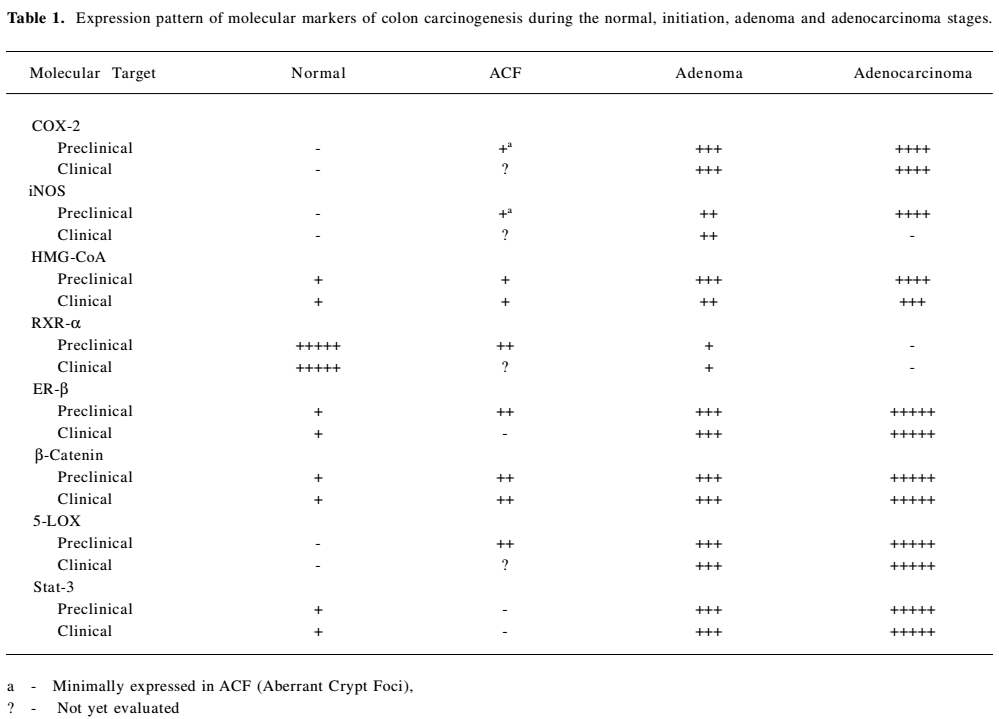
Surrogate endpoint markers provide unique opportunities to understand the cancer development and also to evaluate affectivity of agent’s intervention. All the biomarkers will not achieve the status of surrogate endpoints, only a subset of biomarkers may achieve surrogate endpoint status. Provisional evaluation is not done until it is evaluated in controlled clinical trials or observational investigations (in large populations) for its therapeutic intervention and safety. Based on epidemiologic, therapeutic, pathophysiologic and due to clinical benefit, adenomas are taken as surrogate endpoints in colon cancer. Removal of adenomatous polyps has been shown to reduce the risk of development of colorectal cancer. However, intervention using adenomas as endpoint may not be fruitful, because colonic polyps often take several years to develop and become adenomatous carcinomas. Due to these limitations of adenomas as surrogate biomarkers of cancer, much interest is currently shown on research in the use of surrogate endpoints that are altered early in colorectal carcinogenesis, prior to polyp formation, to predict the clinical effectiveness of chemopreventive drugs. It takes 10–20 years for a normal colonocyte to undergo molecular changes and to be clinically detected as a neoplastic lesion. Rather, ACF (Aberrant Crypt Foci) can be used as endpoint in colon cancer. Aberrant crypts are postulated to be the earliest identifiable potential precursors of colon cancer. Analysis of aberrant crypts may facilitate the study of the early pathological and molecular changes that precede adenoma to colon cancer. The observation of manifold/more aberrant crypts in the mucosa from patients and who are at high risk for colon cancer, compared to the mucosa from autopsy patients without any signs of colon cancer suggests that an increased frequency of these very early lesions predisposes to colon cancer [2,3]. The progression of adenoma to carcinoma is one of the routes to colon cancer. Therefore, aberrant crypt foci may eventually evolve into polyps and, subsequently, cancer. Hence, it provides a simple and economical tool for preliminary screening of potential chemopreventive agents, and it allows a quantitative assessment of the mechanisms of colon carcinogenesis. Changes in biomarkers can reveal how far a lesion has advanced. Based on the mechanism of the drug, alterations in other biomarkers can tell whether a drug has reached the target cells. Restoration of normal levels of the biomarkers by chemopreventive/therapeutic agent can indicate potential response to the intervention by that agent. A drug can act early in the neoplastic pathway to prevent adenoma recurrence, but may not show any effect on large adenomas, so understanding these functions of drugs may help in restoring the altered surrogate biomarkers. As per the biology of cancer cells, like normal cells, most cancer cells use multiple redundant intracellular signaling pathways to ensure the maintenance and viability of functions critical to their survival. Understanding these pathways in detail may help in the identification of such markers. The most important studied/altered molecular markers/targets involved in signal pathways which can be considered under surrogate endpoints in colon carcinogenesis are COX-2, iNOS, HMG-CoA, RXR-α, ER-β, β-catenin, 5-LOX, and STAT3. Effect of different agents on these molecular markers/targets will influence the endpoint markers in colon carcinogenesis.
COX-2 COX-1 and COX-2 are the two isoforms of the COX enzyme [4,5]. COX-1 and COX-2 catalyze the conversion of arachidonic acid to eicosanoids, namely prostaglandins and thromboxanes via endoperoxides. The COX-1 isoform synthesizes prostaglandins that are required for normal physiologic function like gastrointestinal cytoprotection and platelet activity. COX-2 is not detectable in most normal tissues; however, it is induced at sites of inflammation by cytokines, growth factors, tumor promoters and other agents. Both isoforms are also responsible for the synthesis of prostaglandin E2 (PGE2), and there is some evidence for a correlation between increased levels of PGE2 and tumorigenesis [6–8]. Prostaglandins, especially prostaglandin E2, appear to be important in the pathogenesis of cancer secondary to the effects on mitogenesis, cellular adhesion, immune surveillance and apoptosis. PGE2 exerts its actions by binding to one (or a combination) of its four subtypes of receptor (EP1, EP2, EP3 and EP4). Malignant tissues have been found to over-express prostaglandins when compared with normal tissues [9–11]. COX-2 is a key mediator in the development of colon cancer. COX-1 and COX-2 differ in expression and regulation in different tissues. COX-1 is constitutively expressed in the colon, but COX-2 is inducible and markedly up-regulated in many colon cancers [12,13] (Table 1). COX-2 is found to be associated with chronic inflammations. In several animal and human models, the inhibition of prostaglandin formation by blocking COX-2 appeared to show protective effects against many types of cancers, including breast, colon, esophageal, lung, skin and head and neck cancers. Several mechanisms have been proposed to explain the important role of COX-2 in tumorigenesis [14]. Increased COX-2 gene expression has been shown in human colorectal adenocarcinomas and in carcinogen induced rat colonic tumors [12,13,15–17]. Even COX-2 is known to modulate angiogenesis [18,19]. Notable over-expression of COX-2 protein in tumors located in the rectum was observed when compared with other locations in the colon [20]. Certain studies have explained that COX-2 expression is unrelated to overall patient survival [21]. Recent observations of COX-2 expressions with colon tumor samples from patients explain a progressive over-expression of COX-2 during stepwise sequence from adenoma to carcinoma [22]. Still no clear evidence is available on the expression patterns of COX-2 at different stages (I – III) of colon cancer and its effect on progression of carcinogenesis. In depth detailed study is necessary to improve the efficacy of drugs to target COX-2. Due to various evidences of its oncogene actions, suppression of COX-2 is now a crucial target for control of tumors with chronic/persistent inflammation (Figure 1).
Full table
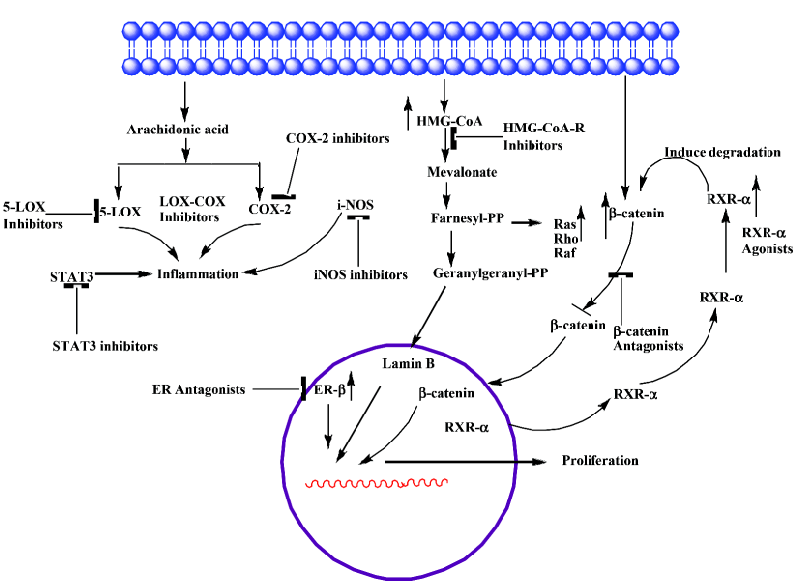
Epidemiological and rodent studies have documented a protective effect of non-steroidal anti-inflammatory drugs (NSAIDs) in preventing colorectal cancer. NSAIDs showed inhibition of COX-2 activity and PGE2 synthesis both in vitro and in vivo [23,24]. We have strong clues from recent findings that non-steroidal anti-inflammatory drugs (NSAIDs), COX-2 inhibitors in particular, are effective chemopreventive agents in colon cancer [25]. The concern over gastric toxicity associated with aspirin (NSAID) use led to efforts to develop COX-2 specific inhibitors (COXibs) [26,27]. Though COXibs are found to be better drugs, several large randomized controlled trials provided unequivocal evidence of the cardiotoxicity of COXibs, which led to withdraw COXibs from US market [28–31] Celecoxib selective COX-2 inhibitor is used to treat FAP and is still in use. Celecoxib is approved as an adjunctive (secondary) treatment among patients with FAP. Celecoxib is found to be effective in combination with DHA (fatty acid) in HCA-7 human colon cancer cells [32]. Different phase (I/II/III) clinical trials are being carried out using celecoxib individually and also in combination with other drugs (curcumin, Docetaxel, prednisolone, Zoledronate, Eflornithine) in colon cancer patients with different case histories to study its efficacy, reduce its toxicity and for better outcome. But there is compelling evidence for the cancer-chemopreventive potential of aspirin and other NSAIDs [22,29,32,33]. A recent report on use of (Classical NSAID) aspirin in men and women with differential expressions of COX-2 showed varied responses on colorectal cancer. Regular use of aspirin appeared to reduce risk in those patients with high levels of COX-2 expression, but there was no effect shown on patients with very low/no COX-2 expression [22]. The differences between colon cancers that express high or low levels of COX-2 have to be investigated, and this understanding can form the basis for clinical implications. This study can likely spur research to identify who is at risk of COX-2-expressing colon cancers and for new drugs that can provide benefits for colon cancer without the risks associated with the drug. Nitric-oxide releasing nonsteroidal anti-inflammatory drugs (NO-NSAIDs) are other promising alternatives to aspirin in chemoprevention of colon carcinogenesis, which possess the beneficial effects of parent compound (aspirin) and at the same time are devoid of side effects [34–36]. NO-aspirin and NO-indomethacin are experimented against the development of colon adenocarcinoma in an established animal tumor model. These agents suppress both invasive and noninvasive adenocarcinomas of the colon [37]. Various mechanistic pathways are suggested for the beneficial effects of NO-NSAIDs [38–40]. Functional studies of prostaglandin-related polymorphisms, including biochemical studies that evaluate the response to NSAIDs and COXibs [41], will provide information on possible subgroups of individuals who might be more susceptible to the toxic effects of either COXibs or standard NSAIDs. NO-NSAIDs and COXibs need to be further evaluated for their eventual use for human treatments.
iNOS Intestinal inflammation is almost invariably accompanied by intestinal dysfunction, which constitutes a major clinical complication. Current therapy is often inadequate due to incomplete knowledge about the mechanisms causing intestinal dysfunction in intestinal inflammation. Nitric oxide (NO) may be a key component in this process. NO is a highly reactive compound that is produced by three isoforms of Nitric Oxide Synthase (nNOS, eNOS and iNOS). Under normal physiological conditions, endogenous NO is produced by the constitutive NOS isoforms, eNOS and nNOS (neuronal NOS). These two are important for peristalsis (nNOS) and maintaining mucosal blood flow (eNOS). NOS (iNOS) is expressed in many cells, extravascular resident leucocytes (macrophages), intravascular and/or infiltrating leucocytes (neutrophils and monocytes), endothelium, and parenchymal cells, including intestinal epithelium after exposure to various inflammatory stimuli (lipopolysaccharide (LPS), tumor necrosis factor (TNF α), or interleukin (IL)-1β. iNOS, produces large amounts of NO for a limited period of time and is an element of the innate immunity. Excessive and prolonged NO production has been suggested to cause intestinal dysfunction in inflammatory bowel disease (IBD) and sepsis. For inflammatory reactions that lead to injury, the role of NO is controversial, with evidence for pro-inflammatory as well as anti-inflammatory effects [42]. NO and its metabolites have a role in mediating many cytotoxic and genotoxic effects [43,44]. Effect of NO on tumor progression is dependent on the type of cells and activity of NOSs. Production of NO and expression of iNOS has often been detected in several established human tumors [45–50] (Table 1).However, considering the physiological role of NO, inhibition of NO generation could have deleterious effects.
It is assumed that chronic inflammation and continuous exposure to NO produced by iNOS leads to neoplastic trans-formation, which is a key step in carcinogenesis. Studies of iNOS indicate that NO produced by iNOS can initiate and/or promote tumorigenesis [51,52]. Mice with mutations in both adenomatous polyposis coli (Apc) and iNos showed fewer adenomatous polyps in the small and large intestines compared with mice with the mutation in Apc alone [53]. iNos-/- mice showed decreased incidence of gastric carcinogenesis that is induced by Helicobacter pylori [54]. These findings identify iNOS as a target for tumor chemoprevention in colon cancer. Indeed, iNOS inhibitors reduced azoxymethane-induced colon cancer and tumorigenesis in Apc-mutant mice and rats [53,55,56]. There is a lot of discrepancy in the role of iNOS in tumour stroma. By contrast, several similar studies produced conflicting results regarding different NOSs and the literature supports both increasing and decreasing NO signaling as a potential strategy. Xu et al demonstrated that delivery of iNOS-over expressing cells to the peritumoural region resulted in increased FAS and FAS-ligand expression, and inhibition of tumor growth in human ovarian cancer and colon cancer xenografts [57]. iNOS by a selective inhibitor, 1400W is found not to have any effect on cells not expressing iNOS at appreciable level. Whereas, it is found to inhibit the growth of human colon cancers as well as of murine breast cancers that express endogenous iNOS [58]. Further studies are needed to know what approach can be applied to balance the activities of various NOSs and target the iNOS in a right way by unraveling the precise mechanisms discrepancies. Presently, both treatment strategies to increasing NO signaling and decreasing NO signaling are being tested. Whereas, pre-clinical studies indicate that NO downregulation might be of value in chemoprevention. Preclinical studies in colon cancer models are encouraging in prevention of tumorigenesis with iNOS selective inhibitors. These studies are in agreement with genetic studies of iNOS knockdown mice but conflicting results in some iNos-/- mouse studies have also been reported [53,54,59,60]. Because of these contradictory findings, more detailed evaluation in preclinical models will be required prior to the clinical evaluation of this strategy, keeping in mind the multiple physiological roles of NO. Whereas, one can trust, based on the overall literature, NO promotes tumorigenesis when associated with chronic inflammation, angiogenesis and the growth of established solid tumors. However, a detailed study is needed to understand NO functions in lymphatic system and in lymph node metastasis. NO interaction between COX-2 signaling and NO signaling is well documented [61], and this observation can be utilized in aiming tumor prevention. A combination of COX-2 inhibitor and iNOS inhibitor has been shown to produce a better chemopreventive effect against colon carcinogenesis [56] (Figure 1). Combinations of anti-angiogenic agents and NOS inhibitors might also be more effective [62]. Exploitation of combination treatments which target multiple targets may be beneficial. Still, there are many gaps to be filled in to efficiently target iNOS.
HMG-CoA reductase HMG-CoA reductase is the key regulated step in cholesterol synthesis and represents the sole major drug target for contemporary cholesterol-lowering drugs. There are conflicting reports in the literature on the association between serum cholesterol level and colorectal cancer. A low level of cholesterol was observed in some studies due to metabolic effect of undiagnosed colon cancer. Positive and negative associations have been observed at different points in time, prior to the diagnosis of cancer. The geographical incidence of colon cancer correlates well with high fat diets [63]. Patients with colon cancer have high levels of faecal bile acids and cholesterol. Results from animal studies also support the significance of fat intake in colon cancer development [64]. It is thought that faecal bile acid and cholesterol metabolites may act as promoters, co-carcinogens or carcinogens in large bowel tumorigenesis. Cholesterol is an obligatory precursor of the bile acid. HMG-CoA reductase is a polytopic, transmembrane protein that catalyzes a key step in the mevalonate pathway [65], which is involved in the synthesis of sterols, isoprenoids and other lipids (Figure 1). These end products are important for many different cellular functions. Acetyl-CoA (citric acid cycle) is converted to acetoacetyl-CoA. Acetyl-CoA condenses with acetoacetyl-CoA to form 3-hydroxy-3methylglutaryl-CoA (HMG-CoA). HMG-CoA is reduced to mevalonate by NADPH. This reaction occurs in the cytosol. It is the committed step in cholesterol synthesis, which is why the enzyme (HMG-CoA reductase) catalyzing the reaction is a target of statins. Blockade of rate limiting step by statins results in reduced levels of mavalonate and its down stream molecules (such as Ras, nuclear lamins, and many small GTP-binding proteins such as members of the Rab, Rac, and Rho families) having significant influences on cellular functions. HMG-CoA reductase is observed to be overexpressed in colon cancer cell lines, which makes it a potential molecular target for colon cancer prevention/treatment [66,67] (Table 1). Statins also exert immunomodulatory and anti-inflammatory effects, which provide them with potential anti-tumor effects [68,69].
HMG-CoA reductase inhibitors such as lovastatin, atorvastatin, pravastatin and simvastatin have been in use for the past 15 years for their efficacy in reducing cardiovascular diseases. These studies suggest merits of statins use in chemoprevention. Whereas, major randomized controlled trials demonstrated no association between the use of HMG-CoA reductase inhibitors and the risk of fatal and nonfatal cancers [70]. A recent large study, the Molecular Epidemiology of Colorectal Cancer (MECC) study, which included almost 4000 people, showed that statin use for a period of more than 5 years was associated with a 47% reduction in the risk of colorectal cancer [71]. Eight previous, small prospective studies found no/weak statistically significant association between statin use and the risk of colorectal cancer and significantly lowered risk with specific statins or their dose levels [72–79]. Although these controversies exist on statins effect on colon cancer, many preclinical data support the positive effects of statins in reducing colon cancer. Different statins were successful in inhibiting colorectal carcinogenesis in rodent models [80–86]. Anti-proliferative effects of HMG-CoA inhibitors on different cancer cell lines are through two cyclin dependent kinases and Rho small GTPases, geranylgeranylated by GGPP [87–90]. Statins are tested positive in combination with cytotoxic drugs, which yield synergism in preclinical models [86,91] Because HMG-CoA reductase inhibitors exhibit diverse effects on various aspects of carcinogenesis in vitro and in vivo, they deserve further investigation in chemoprevention and therapeutic clinical trials. Numerous clinical trials are under way to assess whether these actions will translate into significant clinical benefit. Hence, there is optimism for the use of HMG-CoA reductase inhibitors as anti-neoplastic agents.
RXR-α Retinoids comprise a family of polyisoprenoid lipids consisting of vitamin A and its derivatives. Vitamin A, and its natural and synthetic analogs are found to interact with the RAR and RXR receptors. These compounds exert their multiple functions through these receptors. RXRs are found to have important cross interactions between RXR and other 20 nuclear receptors. RXR forms heterodimers with RAR, VDR, TR, etc. through which they transmit the hormonal signals by interacting with co-activators and co-repressors to regulate genes [92–94]. RXR-α is one of the retinoid X receptors (RXR-α, RXR-β, RXR-γ) and belongs to a member of the nuclear receptor super family which regulates development, organ physiology and cell proliferation. These receptors are altered distinctly in different tissues during carcinogenesis, providing evidence of unique functions of each receptor in different tissues. The expression patterns of these receptors in carcinogenesis provide prognostic information, as these receptors have a prominent role in actions of gene expressions when interacted by a ligand. Among the RAR and RXR receptor subtypes, RXR-α mRNA was observed to be expressed at the highest levels in various normal cell types of gastric mucosa. Due to generally higher levels of RXR-α expression, it is identified to possess more vital functions than other RAR receptor subtypes in normal gastric mucosa. As RXR-α is known to interact with other receptors (Vit D, PPARγ), it may also play a physiological role in the colon. This can be supported by the investigations of Kane et al. that among all the three receptors (RXR-α, RXR-β, RXR-γ) tested, a significant decrease in RXRα expression was observed in human malignant colon tissues, compared with non-neoplastic tissue. RXR-α decreased expression was noted in skin and gastric cancer too [95,96] (Table1). Genetic studies of (RXRα1/-) knockdown mice reported a significantly enhanced susceptibility to 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis compared with their wild-type littermates [97]. RXR has been shown to have a role in decreasing colonic inflammation in mouse models [97]. In addition, RXR-α is found to induce β-catenin proteasomal degradation, replacing the function of APC and shows an interaction of RXR-α with β-catenin [98]. RXR-α may exert chemopreventive or chemotherapeutic effects in the colon via the regulation of β-catenin [98]. Dysregulation of β-catenin is a common outcome of mutations observed in colorectal cancer, resulting in β-catenin induced activation of oncogenes [98]. RXR agonists have been shown to enhance interaction between RXRα and β-catenin, resulting in more efficient β-catenin degradation and subsequent antiproliferative effects [98] (Figure 1). One of the novel functions, RXR-α acts as a carrier for nucleocytoplasmic translocation of orphan receptors [99]. In view of its functions, RXR-α is considered to be an attractive molecular target in colon cancer. Collectively, these results raise the possibility that RXR agonists might have a beneficial role as a therapy in colon carcinogenesis, where activated wnt signaling is believed to contribute to tumorigenesis.
Rexinoids are ligands/agents that selectively bind to retinoid X receptors (RXRs) [100–103]. They are found effective both in prevention and treatment. Most notable rexinoids are bexarotene (LGD1069), LG100268, VTP 194204 studied in other cancers. Bexarotene is the first synthetic RXR-selective agonist to enter for clinical trials for cancer therapy. It is shown to have minimum binding to RARs compared to other rexinoids. It is proved to be effective in preventing ER– and ER+ mammary tumors [104,105]. It is noted that bexarotene is well tolerated without any classiccal signs of traditional retinoid toxicities. Although it is tested for breast cancer, no studies are found on colon carcinogenesis using this compound. LG100268, VTP 194204 are potent selective RXR agonists with no biologically relevant binding to RARs, and have been identified to be more effective than bexaro-tene [106,107], but they needed to be tested in colon cancer animal models before being taken for clinical studies. RXR-specific agonists AGN195362, AGN195456, AGN195741, AGN196060, and AGN196459 were tested with colon cancer cell lines and observed to induce growth inhibition through β-catenin degradation in APC independent manner [98]. This data indicate the role of RXRs in ligand mediated protein degradations in vitro; however, it needs to be validated in animal models. Rather, induction/elevation of RXR-α levels through specific ligands can be an efficient method in treating colorectal cancer. β-ionone is an end ring analog of β-catrotene, which is naturally present in various vegetables. The study conducted in our lab to assess the chemopreventive potential of β-ionone, a naturally-occurring agent, against chemically-induced colon carcinogenesis in F344 rats showed significant suppression of carcinogen-induced colonic ACF formation. In-vitro studies, showed cell growth suppression, induction of apoptosis and also induced RXR-α expression (Unpublished data). Identification of various natural and synthetic drugs selective to RXR-α can be beneficial in prevention/treatment of colon carcinogenesis.
ER-β ER-β and ER-α nuclear receptor transcription factors are two forms of estrogen receptors which have distinct expression patterns in human tissues [108]. Estrogens (estradiol, estriol and estrone) are the steroid hormones which exert their actions through these receptors. ER is also found attached to the cell membrane and is involved in signaling by forming complexes with G proteins, striatin, receptor tyrosine kinases (eg EGFR and IGF-1) and non-receptor tyrosine kinases and Cav-1 [109–112]. Several epidemiologic studies have suggested that CRCs are influenced by steroid hormones [113–116]. Endogenous estrogen production is liked to enhancement of growth and metastases of CRCs in female patients [117]. Estrogen Replacement Therapy studies explain the protective effect on colon cancer risk, whereas in these studies, a mostly protective effect is shown by the progesterone rather than estrogen [118–122]. Recent re-analysis of world wide data on the relationship between hormone replacement therapy and breast cancer showed that the risk of this cancer may be increased in women who are taking estrogen [123]. Estrogen receptors are reported to be present in colorectal tissue and colorectal adenocarcinoma and ER-β is found to be the predominate receptor expressed in colonic tissue [124–130] (Table 1).
Various reports suggest presence of five different isoforms of ER-β in colon and colon cancer, due to which, the complexity to determine ER mediated functions has increased. Estradiol treatment was found to significantly enhance the growth of colon cancer cells injected in mice, and the resulting tumors were notably larger in female than in male mice [131]. As most of the actions of estrogens seem to be exerted through the respective receptors [132,133], it suggests that the estrogen receptor may be important in regulating colorectal cancer risk associated with these hormones [134]. Most of the reported data on colorectal cancer suggest the changes in expression patterns of ER-β, rather than ER-α, which point that ER-β gene to be more important than ER-α in the etiology of colorectal cancer [135–139]. ER-α is certainly associated with more differentiated tumors in breast and other cancers, while the evidence that ER-β is involved is controversial. ER-β protein levels are reportedly lower in colon tumors than normal colon tissue, and loss of ER-β is linked to its protective role [135–139]. Whereas, certain reports suggest that increase in levels of ER-β in colon tumorigenesis is associated with advanced stages of colon cancer and tumor cell differentiation [140]. ERβ-/- knock out mice studies reported a role of ER-β in the organization and architectural maintenance of the colon, whereas it could not prove that an exact lack of ER-β is causing hyperproliferation, as the cells were lost due to shedding [141]. Hence, ambiguity on the protective role of ER-β still exits.
Taken the differential functions of estrogen receptors into consideration, a class of drugs termed “selective estrogen receptor modulator” (SERMs) were developed (Figure 1). The concept of selective estrogen receptor modulator is based on the ability to selectively activate (or block) one type of ER or to promote ER interactions with different proteins, such as transcriptional co-activator or co-repressor proteins. Additionally, the different estrogen receptor combinations respond differently to various antagonists, and some compounds have partially agonistic and antagonistic effects, depending on the tissue [142]. One such antiestrogens or SERMs which is used to inhibit the growth of colon cancer and is widely used is anti estrogen tamoxifene, which has shown good application in colon cancer cells and is also efficient in inhibiting liver metastates from CRCs in animal models [143]. Whereas, the effect of tamoxifene on CRC in different observational and clinical studies were not encouraging either, there was no effect or there was a modest increase/increased risk of CRC [144–146]. Due to tamoxifene associated side effects, another SERM, raloxifene, is tested for its efficacy in cancers and has been approved by FDA for prevention of osteoporosis. It is found to be safe when used as a preventative chemotherapy for women judged to have a high risk of developing breast cancer or who have breast cancer [147,148]. The accumulated results from MORE, CORE and RUTH studies suggest that raloxifene use is safe and does not appear to increase the risk of CRC [149,150]. Raloxifene has shown to have biological effects on CRC ERâ positive cell lines [151]. Our observations with raloxifene on colon cancer prevention showed good inhibition of AOM induced tumors in rats and growth inhibition of colon cancer cell lines (Data not yet published). Droloxifene, idofene and toremifene are similar SERM agents, but they still need to be experimented to be considered. Another chemotherapeutic anti-estrogen, ICI 182,780 (Faslodex), which acts as a complete antagonist, also promotes degradation of the estrogen receptor, even this can be considered experimental in colon cancer prevention. ER-β isoforms predominate in the colon and each isoform needs to be evaluated for its ligand dependent and independent effects on cell growth, development or death, as the physiological significance of these ER isoforms is still unknown [152,153]. Valuable information can be achieved by expanding knowledge on estrogen effects by determining the ER-β expression in normal colon and cancerous tissues in patients (men and women) below 40 years and also of ER-β isoforms association with different responses to estrogens and anti-estrogens. This knowledge may help in the better designing of drugs to counteract the progression of colon carcinogenesis.
β-catenin β-catenin is a subunit of the cadherin protein complex. β-catenin is found at the plasma membrane in association with cadherins, in association with the tumor suppressor promoter APC and microtubules, in the cytoplasm and in the nucleus. So, it cannot be presumed that it works only in the nucleus to “signal” a response, it is a multifunctional protein involved in cell adhesion, signaling and many more [154,155]. β-catenin is shown to regulate pre-mRNA splicing [156]. There are a number of reports pertaining to the functional interactions between nuclear receptors and the canonical, Wnt/β-catenin signaling pathway cascade [157]. β-catenin seems to interact with various nuclear receptors and it is shown that genetic interactions between ER and β-catenin may promote growth and tumorigenesis in eye of drosophila [157,158]. The significance of this data remains to be experimented in other systems to give a better idea of the functions and interactions of β-catenin with nuclear receptors. The deregulation of β-catenin has been found to lead to various forms of cancer, particularly colorectal cancer [159]. Normal function of APC in combination with glycogen synthase kinase 3β and axin regulates free cytoplasmic β-catenin levels by binding to and targeting β-catenin for degradation by ubiquitination-dependent proteolysis [160–165]. This regulates the availability of free β-catenin for binding with the TCF-LEF family of transcription factors in the nucleus [166]. Mutations in APC or β-catenin can result in the failure of β-catenin to be degraded and they are retained in the cytoplasm of cells which is often seen in colon cancer [159,167–169] (Table 1). Subsequently, there will be an increase in β-catenin-TCF complex formation, causing alterations in gene transcription (myc, cyclinD1, c-jun, Tcf-1, Lef-1, conductin/axin2 MMP7), leading to carcinogenesis [170,171]. Somatic mutations in genes in the β-catenin pathway are found in >80% of colon cancers (FAP and sporadic disease), and aberrant beta catenin activity is known to play an early and causative role in colon cancer. This may occur by mutations in the APC protein, axin or in the β-catenin itself, which leads to dysregulation of β-catenin turnover and activation of genes involved in tumorigenesis [166,167]. As these mutations are exclusively found in colorectal cancers, β-catenin is considered to be a potential molecular target in colon carcinogenesis [170,171]. A study with folic acid explains that certain dietary supplements can enhance the tumor formations through β-catenin accumulations. Dietary supplementation of folic acid is found to enhance colorectal carcinogenesis through a distinct APC mutated pathway [174] and this explains that folate can act as tumor inhibitor only under particular settings with a specific genetic status of the disease. A recent publication from an outcome of clinical study also supports the findings that folic acid did not show any effect in preventing colon cancer rather it was observed to enhance colon cancer [175]. May be more specific study involving the patients having colon tumors with specific genetic aberrations can help in completely understanding the role of folic acid.
Previous results demonstrated that PPAR-gamma and RXR-alpha may interact with and stabilize a beta-catenin transcription complex in some colon malignant cells. The interaction of ligand, such as NSAIDs with PPAR-gamma may induce the conformational change of the receptor, leading the inhibition of transactivation function of beta-catenin, finally blocking Wnt/beta-catenin signaling [176]. Reports suggest the interaction of RXR receptors with β-catenin induce degradation of β-catenin in colon cancer cell lines [98]. This mechanism may have a significant value to the development and selective use of rexinoids and NSAIDs-like drugs as cancer chemopreventive and chemotherapeutic agents. A number of compounds that inhibit the beta-catenin pathway and show selective toxicity toward cancer cells have been identified. Currently, there are over 20 anti-sense oligonucleotides in clinical trials [172,177]. Diverse β-catenin antagonists have been developed [173,176,178] (Figure 1).The most advanced compounds, the PRLX 8025 compound series, are currently in pre-clinical efficacy testing in animal models of colon cancer [179]. The development of small molecules which selectively target beta catenin interaction that transmits the cell proliferative signal of beta catenin, while leaving alone the other activities of beta catenin required for normal cell growth, may be a viable option for treatment and prevention. The development of drugs that selectively target the nuclear entry or exit of regulatory proteins altered in cancer may be another option for treatment and prevention.
5-LOX Lipoxygenase (LOX) is one of the two important enzyme classes that metabolize polyunsaturated fatty acids and affect carcinogenesis. Cell membrane phospholipids are converted to arachidonic acid, which serves as a substrate that gives rise, in turn, to two powerful and potentially damaging classes of inflammation mediators, known as eicosanoids: the prostaglandins and leukotrienes. Arachidonic acid release and production of eicosanoids are prerequisites for inflammation. The inflammatory prostaglandins and leukotrienes are formed by the action of cyclooxygenase (COX-2) and lipoxygenase (5-LOX) enzymes, respectively [180]. This forms the crux of dual inflammatory pathways: COX-2 and 5-LOX. Many reports documented the clear evidence of COX pathway generating inflammatory prostaglandins and its role in colon carcinogenesis. But medical research has largely ignored the potentially damaging effects of 5-LOX, the enzyme that forms the second branch of the dual arachidonic acid inflammation pathways. Most emphasis was given to block COX-2 activity, ignoring the effects of 5-LOX, which may actually increase the 5-LOX levels, worsening the inflammation. This may be due to shifting of arachidonic acid toward synthesis of leukotrienes through the 5-LOX pathway when COX-2 is blocked, which plays a vital role in inflammation. COX-2 inhibition alone was ineffective in slowing the progression of clinically diagnosed cancers. It is clearly evident from animal models and in vitro studies that expression of 5-LOX appears to be occasionally upregulated during neoplastic transformation [181–183] (Table 1). Expression of 5-LOX has been characterized in early colon neoplasms and found upregulation of 5-LOX in colon polyps and colon cancer [184–187]. These observations provide evidence that 5-LOX plays a role in colon cancer development and may be an early target for chemoprevention of colon cancer.
Leukotrienes are involved in cell survival signals which indicate proliferative properties of a cell [188]. 5-LOX is observed to be involved in exerting both anti and pro inflammatory activities. The other bioactive compounds which need to be studied further to understand the network of anti and pro inflammatory mediators are lipoxins and resolvins. Biosynthesis of LO-derived eicosanoids (Lipoxins) occurs through different pathways. One of the pathways involves 5-LOX in the formation of metabolically active products (lipoxins), LXA4, LXB4, or 15(R)-HETE for 15-epi-LXs (aspirin triggered LXs) (LTA4/LTB4). These lipoxins are known for promoting the resolution of inflammation and also development of tumors. Biologically active metabolites of the 5-LOX cascade are LTB4 and the so-called cysteinyl LTs (LTC4, LTD4, and LTE4), which are proinflammatory mediators. Cysteinyl leukotrienes (CysLT) are important proinflammatory mediators, which exert effects on several cellular functions, including smooth muscle contraction, bronchial mucus production and chemotaxis [189]. Recently, it has been shown that even these products may contribute to the development of colon and several other human tumors [188,190,191]. Lipoxins are high affinity antagonists to the cystienyl leukotriene receptor 1 (CysLT1) to which several leukotrienes (LTC4, LTD4 and LTE4) mediate their smooth muscle contraction and eosinophil chemotactic effects. Also, pharmacological evidence provides the modulation of CysLT1 mediated inflammatory processes in vivo by antiinflammatoty lipids [192]. 5-LOX is even involved in E series resolvins (anti-inflammatory), suggesting its protective role in addition to its leukotriene production. These results are confirmed with recent reports of exacerbated inflammatory responses in 5-LOX deficient mice [193,194]. These findings are contradictory and explain for a further insight into actual underlying mechanisms and the networks involved during inflammation and also at the resolution of inflammation, as these lipoxins are formed during the resolution of inflammation process. 5-LOX protective mediation is through lipoxins and resolvins. As 5-LOX is not the only pathway for the production of these anti-inflammatory components, use of 5-LOX inhibitors can prove beneficial (Figure 1). Thus, as a therapeutic approach, inhibitors of 5-LOX may come without many side effects. Each of these proinflammatory mediators activates specific signaling mechanisms. The potential role of lipoxins and resolvins in preventing chronic inflammation needs to be investigated. The use of experimental animal models in colon cancer will help to underpin the relative importance of lipoxins and resolvins in colon carcinogenesis. Data available on Lipoxins and resolvins as promoting the resolution of inflammation urge the need to develop agonists of lipoxins and resolvins. Nevertheless, drugs able to block the lipoxygenase pathway, 5- lipoxygenase inhibitors or leukotrienes receptor antagonists, although they are included among the effective therapies of asthma, appear to be an insufficient single therapeutic approach of inflammation. It is likely that these results reinforce a growing body of research that dual inflammatory pathway inhibition may be needed to fully realize the promise of anti-inflammatory therapy [195].
The class of dual 5-Lox/Cox inhibitors such as Indometacin, dexamethasone, ER-34122, BW 755C, diclofenac, Tepoxalin and licofelone has emerged as an effective and well tolerated therapy that could offer safety advantages over Cox inhibition alone [196]. These dual inhibitors are tested and found to be effective in animal models and clinical trials [197,198]. Tepoxalin was proved to be better than indomethacin in having no gastro intestinal toxicity [199 –201]. Based on various observations, Tepoxalin have future prospects as preventive and therapeutic drug in colon carcinogenesis. Licofelone is a novel dual 5-LOX/COX inhibitor today in phase III clinical development that effectively inhibits the synthesis of cysteinyl LTs [197,202,203]. The observations of the clinical development of licofelone indicate that this drug also has an excellent GI profile.
STAT-3 Stat (signal transduction and translation) proteins, normal constituents of cells activated by tyrosine kinases, have diverse biological functions [201]. Any aberrations in stat signaling are predicted to have a wide variety of consequences. Stat 1, 3 and 5 are observed to be more strongly associated with human cancers out of the known stat family members. Although dysregulated activity of STAT factors 1, 3 and 5 has been implicated in various cancers, not many studies have addressed these in the context of CRC. Experimental and clinical data have revealed the oncogenic potential of STAT3 through over expression and constitutive activation in a variety of human malignancies, including leukemia, melanoma, head and neck squamous cell carcinoma, breast, prostate, ovarian and colorectal carcinoma [198,204 –209] (Table 1). STAT3 is activated by cytokine and growth factor receptors, several viral or cellular oncogenes, such as src, fps, polyoma virus middle T-antigen and sis [205,210]. Cell transformation by aberrant STAT3 activity involves upregulation of genes promoting cell cycle progression (cyclin D1 and c-myc) and/or preventing apoptosis (bcl-xL, mcl-1, and survivin) [211–214]. These findings make it a suitable target for disease intervention. It is presumed that STAT3 elicits permanent changes in the genetic constitution of the cell required for the initiation and maintenance of tumorigenesis. STAT3 plays a vital role in tumor angiogenesis by acting as a direct transcriptional activator of VEGF [215,216]. It is identified that STAT3 is activated by modulation of both tyrosine phosphorylation and lysine acetylation to stimulate cancer cell growth and metabolism. These findings help in better targeting of STAT3. One can target upstream (JAK, SRC, EGFR) and downstream targets responsible for activation of STAT3. The results of STAT3 disruption in mouse fibroblasts demonstrate that deletion of STAT3 is not deleterious for normal functioning of cells [217,218]. This observation clears the fears of drastic effects to be normally associated by blocking STAT3 functioning. The molecular mechanism of oncogenesis by STAT3 is yet to be defined in colon carcinogenesis. STAT3 protein is involved in mediating cytokine signaling, which in turn, has a vital role in inflammation of cells. It is known in colon cancer that inflammatory cells have powerful effects during the early neoplastic process, these cells promote tumor development, facilitate genomic instability and promote angiogenesis. By these actions of activated STAT3, one might assume that it is playing a vital role in colon carcinogenesis. Genetic approaches and also small molecule inhibitors (antitumor cytokines) might prove to be better approaches for inhibiting activated STAT3. Efficacy studies in animal models are to be carried out to screen various naturally occurring anti-inflammatory molecules which have a promise in inhibition of activated STAT-3 and further prevention of colon carcinogenesis (Figure 1). The most promising compounds from animal models relevant to human diseases showing no toxicities can be studied further in human clinical trials.
siRNAs
Small interfering RNAs (siRNAs) are tiny bits of genetic material that can prevent the translation of genes into proteins — including specific proteins involved in biochemical reactions that promote cancer and other diseases. siRNA interference has emerged as an invaluable tool in biological research, and could also become a powerful therapeutic modality because of its broad applicability, specificity and high efficiency [219–221]. There are many reports on use of siRNA in silencing a gene of interest in many different diseases. including cancers [222]. siRNA approach was found effective against TGFβ1 and reduced its expression in HCT-116 colon cancer cells [223]. Treatment with a COX-2 siRNA in colon cancer cells demonstrated a significant knockdown of COX2 at the protein level of 57%, as compared to a non-silencing siRNA control [224]. Down-regulation of endogenous levels of COX-2 can be achieved in colon cancer by siRNA. This strategy should prove to be a valuable tool in revealing the specific function of COX-2 in tumorigenesis [224]. RNA-i based technologies have gained popularity by addressing the ability to knockdown several genes at the same time, but this can be a disadvantage if considered a specific knockdown. siRNA was used to obtain defined combinations of pro- and antiapoptotic gene expression in colon cancer cells of varying p53 status [225]. siRNA is also used in elucidating functional role of LPA (Lysophosphatidic acid) and LPA receptors in colorectal cancer cells [226]. These observations demonstrate Bcl-2 and LPA receptors accessibility for siRNA silencing, making specific gene targeting a very possible approach. siRNAs targeted against different cancer related pathways in colon carcinogenesis are tested in mice successfully, whereas there are many hurdles needed to be cleared before it can be implemented in human treatments. It can be developed as a new treatment tool, provided it can be delivered to the site of interest, and is able to be assessed through non-invasive methods, specific siRNA sequence and mode of delivery. There are possible toxicities induced by partial inhibition of homologous genes/function of endogenous miRNAs. If one can overcome the problems, this can become an attractive therapeutic model for colon carcinogenesis.
Micro RNAs–emerging new science as a target for multiple diseases
MicroRNAs (miRNAs) are short non-coding RNA molecules that regulate expression of genes by repressing translation or by cleaving RNA transcripts [227,228]. These play a crucial role in cell differentiation/development, proliferation and apoptosis [229]. Therefore, they are believed to play an important role in cancer development and can become potential therapeutic targets. Each miRNA can target hundreds of transcripts directly or indirectly [227–230], whereas more than one miRNA can converge on a single mRNA target [231–235]. Recent studies showed that miRNAs have been deregulated in various cancers including colon [236–239]. The expression profiles of miRNAs can be used for the classification, diagnosis and prognosis of human malignancies [240]. miRNA expression is frequently disregulated in cancer cells, and specific miRNAs are known to regulate both cell-cycle progression and apoptosis [241–243]. A recent report demonstrates that miRNAs are important components of the p53 transcriptional network in HCT116 colon adenocarcinomas cells. Their observations suggest an important role for miR-34a in mediating p53 tumor suppressor function [244]. Mutations in miRNAs are observed in breast cancer tissue samples and the role of mutations in miRNAs is still to be elucidated [240]. A number of molecular studies have shown that colon carcinogenesis results from an accumulation of epigenetic and genetic alterations, including activating and inactivating mutations. This suggests that alternative genetic events may occur during colorectal carcinogenesis. Still, there is no clear understanding in relating all these events at the molecular level in colon cancer development. It is known from a few reports that colon cancer is associated with altered expression of mi-RNAs. Michael et al reported that the expression of miR-143 and -145 was consistently reduced at the adenomatous and cancer stages of colorectal cancer [245]. Later, Bandres et al reported the most significantly deregulated mi- RNAs were miR-31, miR-96, miR-133b, miR-145 and miR-183 by studying the expression of 156 mature miRNAs in a panel of 16 colorectal cancer cell lines and 12 matched pairs of tumor and non-tumor tissues from patients [239]. In addition, the expression level of miR-31 was correlated with the stage of colorectal cancer. Yukihiro Akao et al, have shown MAPK transduction proteins, such as MAP3K and MAP4K4, are possible targets for miR-145 [246]. Because up to hundreds of target genes may be affected by a single miRNA, as predicted by bioinformatics approaches and thus, a given miRNA may target several hundreds of miRNAs that would include transcripts of oncogenic or anti-oncogenic genes, so, it may be difficult to determine the oncogenic functions of a given miRNA in cancer cells. Rapidly emerging information on miRNAs as a new science, which is known to regulate a vast number of genes in maintaining normal crypt, a study is necessary to identify specifically those altered miRNAs related to specific pathways and to target them to restore the normal functioning of the colon cells. Further understanding of the biological and functional mechanisms of miRNA is needed to know how miRNA contributes to carcinogenesis. Targeting miRNAs could provide an important diagnostic for prevention/ therapeutic strategy for human colorectal cancer in the future.
piRNAs
Piwi interacting RNAs (piRNAs) are a new class of small non-coding RNAs that possess unique long length of nucleotides (26 – 31), which differ from other miRNAs and siRNAs. These are named as piRNAs due to their association with Piwi proteins, which belong to Argonaute proteins. These are found to be present in sperm-producing cells in mammals. These piRNAs are present during the initiation of meiosis of sperms and observed to disappear by the time sperm matures. Hence, it is suggested that these may regulate germ cell maturations. It is also said that piRNAs have a possible role as a type of immune system against transposons. piRNAs role in cancer is yet to be elucidated. Whereas, hiwi gene (a human member of the piwi family), is known to cause germ cell malignancy [247]. Also, expression of hiwi in human gastric cancer is associated with proliferating cells [248]. These correlations may raise the questions of whether these piRNAs are related to expression of piwi proteins in relation to cancers. piRNAs are not yet studied in relation to colon carcinogenesis. Very little has been reported about piRNAs, but the research is occurring at such a speed that a lot more information may be reported giving many clues by the time this article appears.
Conclusions
The ultimate goal of chemoprevention is the reduction of cancer incidence by intervening developmental pathways in tumor cells which promote growth and metastases of the tumor. Tumor cells develop/possess multiple pathways of survival, whereas identification of a particular group of individuals with alterations in specific pathway and targeting that pathway is a valid approach. Screening and development of agents which have potential preventive properties with multi-target suppression is appreciable. It is important to determine the non toxic dose by elucidating the mechanisms by which the drug acts in in vivo models. Clinical studies are needed to determine whether supplementation of other molecules to reduce the known toxicity of that drug reliably rescues from the induced toxicity. As chemoprevention involves administering nontoxic agents to otherwise healthy individuals who may be at increased risk for cancer, agents with no/minimal temporary side effects have promise of those being used in chemotherapy. Observations of alterations in a particular pathway in specific cancers and identification of risk groups with aberrations in a particular gene are exploited for treatment/chemotherapy. NSAIDs are particularly effective in patients showing COX-2 expression in preventing colon cancer, likewise, statin’s are found efficient in cholesterol related carcinogenesis in which HMG-CoA reductase is a known target. Especially inflammatory related molecular targets are exploited by COX/LOX inhibitors, iNOS inhibitors. It is observed that hormonal receptors are playing a potential role in promoting/inhibiting colorectal carcinogenesis for which ER-β antagonists (SERMs) and RXR agonists (rexinoids) are employed respectively. Overall these drugs are shown to effectively degrade β-catenin, an important biomarker in colon cancer, even though oligonucleotides specific against β-catenin are under investigations for clinical implementation. As most of these pathways are interlinked, combination modalities hold promise for prevention/treatment of colon cancer. It is very obvious from available data that chronic inflammation is a key for colorectal carcinogenesis which leads to alterations in various molecular biomarkers which can be exploited in chemoprevention/therapy.
The emerging new fields of siRNA, miRNAs and piRNAs can be used in combination with selective chemotherapeutic agents in treating colon cancer. There is hope for developing new agents/strategies for chemotherapy. In fact, new colorectal cancer cases occur each year in 21% out of 1444920 men and women in the United States. If chemoprevention can prevent one or even 1000 occurrences of cancer without complications, it can be deemed successful. Chemoprevention has the potential of providing an important means for cancer risk reduction.
Acknowledgements
We extend our thanks to Ms Alyson ATCHISON for editing this article.
References
- O’Shaughnessy JA, Kelloff GJ, Gordon GB, Dannenberg AJ, Hong WK, Fabian CJ, et al. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clin Cancer Res 2002;8:314-46.
- Pretlow TP, O’Riordan MA, Pretlow TG, Stellato TA. Aberrant crypts in human colonic mucosa: putative preneoplastic lesions. J Cell Biochem Suppl 1992;16:55-62.
- Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, et al. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med 1998;339:1277-84.
- Herschman H. Prostaglandin synthase 2. Biochim Biophys Acta 1996;1299:125-40.
- Spencer AG, Thuresson E, Otto JC, Song I, Smith T, DeWitt DL, et al. The membrane binding domains of prostaglandin endoperoxide H synthases 1 and 2. Peptide mapping and mutational analysis. J Biol Chem 1999;274:32936-42.
- Smith WL. Prostanoid biosynthesis and mechanism of action. Am J Physiol 1992;263:181-91.
- Lee SH, Soyoola E, Chanmugam P, Hart S, Zhong H, Liou S, et al. Selective expression of mitogen inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem 1992;267:25934-8.
- O’Neill GP, Ford-Hutchinson AW. Expression of RNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett 1993;330:156-60.
- Marnett LJ. Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res 1992;52:5575-89.
- Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med 1993;122:518-23.
- Giovannucci E, Egan KM, Hunter DJ, Stampfer MJ, Colditz GA, Willett WC, et al. Aspirin and the risk of colorectal cancer in women. N Engl J Med 1995;333:609-14.
- Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase-2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994;107:1183-8.
- Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res 1995;55:3785-9.
- Aurelie T, Christophe F, Nicole P, Jean-Pierre H. Mechanisms leading to COX-2 expression and COX-2 induced tumorigenesis: topical therapeutic strategies targeting COX-2 expression and activity. Anticancer Agents Med Chem 2006;6:187-208.
- DuBois RN, Radhika A, Reddy BS, Entingh AJ. Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology 1996;110:1259-62.
- Kutchera W, Jones DA, Matsunami N, Groden J, McIntyre TM, Zimmerman GA, et al. Prostaglandin H synthase 2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proc Natl Acad Sci USA 1996;93:4816-20.
- Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA 1997;94:3336-40.
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998;93:705-16.
- Masferrer JL, Kathleen ML, Alane TK, Ben SZ, Steven LS, Mark BW, et al. Antiangiogenic and antitumor activities of cyclooxygenase 2 inhibitors. Cancer res 2000;60:1306-11.
- Dimberg J, Samuelsson A, Hugander A, Söderkvist P. Differential expression of cyclooxygenase 2 in human colorectal cancer. Gut 1999;45:730-2.
- Richard F, Matthias S, Klaus-Peter T, Christoph HG, Peter F. Cyclooxygenase-2 Expression in Human Colorectal Cancer Is Unrelated to Overall Patient Survival. Clin Cancer Res 2005;11:4754-60.
- Chan AT, Ongino S, Charles SF. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med 2007;356:2131-42.
- DuBois RN, Smalley WE. Cyclooxygenase, NSAIDs and colorectal cancer. J Gastroenterol 1996;31:898-906.
- Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 1997;99:2254-9.
- Rayyan Y, Williams J, Rigas B. The role of NSAIDs in the prevention of colon cancer. Cancer Invest 2002;20:1002-11.
- Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 2001;1:11-21.
- Dubé C, Rostom A, Lewin G, Tsertsvadze A, Barrowman N, Code C, et al. The use of aspirin for primary prevention of colorectal cancer: A systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med 2007;146:365-75.
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 2005;352:1092-102.
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352:1081-91.
- Solomon SD, McMurray JJV, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005;352:1071-80.
- Rostom A, Dubé C, Lewin G, Tsertsvadze A, Barrowman N, Code C. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the US Preventive Services Task Force. Ann Intern Med 2007;146:376-89.
- Swamy MV, Cooma I, Patlolla JM, Simi B, Reddy BS, Rao CV. Modulation of cyclooxygenase-2 activities by the combined action of celecoxib and decosahexaenoic acid: Novel strategies for colon cancer prevention. Mol cancer Ther 2004;3:215-21.
- DuBois RN. New paradigms for cancer prevention. Carcinogenesis 2001;22:691-2.
- Rigas B, Kashfi K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol Med 2004;10:324-30.
- Yeh RK, Chen J, Williams JL, Baluch M, Hundley TK, Rosenbaum RE, et al. NO-donating nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit colon cancer cell growth more potently than traditional NSAIDs: a general pharmacological property? Biochem Pharmacol 2004;67:2197-205.
- Kashfi K, Borgo S, Williams JL, Chen J, Gao J, Glekas A, et al. Positional isomerism markedly affects the growth inhibition of colon cancer cells by nitric oxide-donating aspirin in vitro and in vivo. J Pharmacol Exp Ther 2005;312:978-88.
- Rao CV, Reddy BS, Steele VE, Wang C-X, Liu X, Ouyang N, et al. Nitric oxide–releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Mol Cancer Ther 2006;5:1530-8.
- Kashfi K, Ryan Y, Qiao LL, Williams JL, Chen J. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect. J Pharmacol Exp Ther 2002;303:1273-82.
- Gao J, Liu X, Rigas B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc Natl Acad Sci USA 2005;102:17207-12.
- Hundley TR, Rigas B. Nitric oxide-donating aspirin inhibits colon cancer cell growth via mitogen-activated protein kinase activation. J Pharmacol Exp Ther 2006;316:25-34.
- Fries S, Grosser T, Price T, Lawson J, Kapoor S, DeMarco S, et al. Marked interindividual variability in the response to selective inhibitors of cyclooxygenase-2. Gastroenterology 2006;130:55-64.
- Bogdan C. Nitric oxide and the immune response. Nat Immunol 2001;2:907-16.
- Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumor progression. Lancet Oncol 2001;2:149-56.
- Wink DA, Vodovotz Y, Cook JA, Krishna MC, Kim S, Coffin D, et al. The role of nitric oxide chemistry in cancer treatment. Biochemistry 1998;63:802-9.
- Chhatwal VJ, Ngoi SS, Chan ST, Chia YW, Moochhala SM. Aberrant expression of nitric oxide synthase in human polyps, neoplastic colonic mucosa and surrounding preitumoral normal mucosa. Carcinogenesis 1994;15:2081-5.
- Ambs S, Merriam WG, Bennett WP, Felley-Bosco E, Oqunfusika MO, Osen SM, et al. Frequent nitric oxide synthase 2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res 1998;58:334-41.
- Ambs S, Bennett WP, Merriam WG, Ogunfusika MO, Oser SM, Harrington AM, et al. Relationship between p53 Mutations and Inducible nitric oxide synthase expression in human colorectal cancer. J Natl Cancer Inst 1999;91:86-8.
- Kojima M, Morisaki T, Tsukahara Y, Uchiyama A, Matsunari Y, Mibu R, et al. Nitric oxide synthase expression and nitric oxide production in human colon carcinoma tissue. J Surg Oncol 1999;70:222-9.
- Fransen K, Dimberg J, Osterström A, Olsson A, Söderkvist P, Sirsjö A. Nitric oxide synthase 2 mRNA expression in relation to p53 and adenomatous polypsis coli mutations in primary colorectal adenomas. Surgery 2002;131:384-92.
- Moochhala S, Chhatwal VJS, Chan STF, Ngoi SS, Chia YW, Rauff A. Nitric oxide synthase activity and expression in human colorectal cancer. Carcinogenesis 1996;117:1171-4.
- Hofseth LJ, Hussain SP, Wogan GN, Harris CC. Nitric oxide in cancer and chemoprevention. Free Radic Biol Med 2003;34:955-68.
- Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol Cancer Ther 2003;2:815-23.
- Ahn B, Ohshima H. Suppression of intestinal polyposis in ApcMin/+ mice by inhibiting nitric oxide production. Cancer Res 2001;61:8357-60.
- Nam KT, Oh SY, Ahn B, Kim YB, Jang DD, Yang KH, et al. Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 2004;53:1250-5.
- Rao CV, Kawamori T, Hamid R, Reddy BS. Chemoprevention of colonic aberrant crypt foci by an inducible nitric oxide synthase-selective inhibitor. Carcinogenesis 1999;20:641-4.
- Rao CV, Indranie C, Simi B, Manning PT, Connor JR, Reddy BS. Chemopreventive properties of a selective inducible nitric oxide synthase inhibitor in colon carcinogenesis, administered alone or in combination with celecoxib, a selective cyclooxygenase-2 inhibitor. Cancer Res 2002;62:165-70.
- Xu W, Liu L, Charles IG. Microencapsulated iNOS-expressing cells cause tumor suppression in mice. FASEB J 2002;16:213-5.
- Thomsen LL, Scott JMJ, Topley P, Knowles RG, Keerie AJ, Frend AJ. Selective inhibition of inducible nitric oxide synthase inhibits tumor growth in vivo: studies with 1400W, a novel inhibitor. Cancer Res 1997;57:3300-4.
- Scott D, Hull MA, Cartwright EJ, Lam W, Tisbury A, Poulsom R, et al. Lack of inducible nitric oxide synthase promotes intestinal tumorigenesis in the ApcMin/+ mouse. Gastroenterology 2001;121:889-99.
- Hussain SP, Trivers GE, Hofseth LJ, He P, Shaikh I, Mechanic LE, et al. Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res 2004;64:6849-53.
- Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 2005;310:1966-70.
- Tozer GM, Prise VE, Wilson J, Cemazar M, Shan S, Dewhirst MW, et al. Mechanisms associated with tumor vascular shut-down induced by combretastatin A-4 phosphate: intravital microscopy and measurement of vascular permeability. Cancer Res 2001;61:6413-22.
- Järvinen R, Knekt P, Hakulinen T, Rissanen H, Heliövaara M. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br J Cancer 2001;85:357-61.
- Lipkin M, Yang K, Edelmann W, Xue L, Fan K, Risio M. Preclinical mouse models for cancer chemoprevention studies. Ann N Y Acad Sci 1999;889:14-9.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990;343:425-30.
- Wachtershauser A, Akoglu B, Stein J. HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2. Carcinogenesis 2001;22:1061-7.
- Hentosh P, Yuh SH, Elson CE, Peffley DM. Sterol-independent regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in tumor cells. Mol Carcinog 2001;32:154-66.
- Mason JC. Statins and their role in vascular protection. Clin Sci (London) 2003;105:251-66.
- Crisby M. Modulation of the inflammatory process by statins. Drugs Today (Barcelona) 2003;39:137-43.
- Bjerre LM, Lelorier J. Do statins cause cancer? A meta-analysis of large randomized clinical trials. Am J Med 2001;110:716-23.
- Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, et al. Statins and the risk of colorectal cancer. N Engl J Med 2005;352:2184-92.
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 1996;335:1001-9.
- Downs JR, Clearfield DO, Weis S, Whitney E, Shapiro D, Beere P, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. JAMA 1998;279:1615-22.
- Blais L, Desgagne A, LeLorier J. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and the risk of cancer: a nested case-control study. Arch Intern Med 2000;160:2363-8.
- Probstfield JL, Davis BR. Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care. JAMA 2002;288:2998-3007.
- Graaf MR, Beiderbeck AB, Egberts ACG, Richel DJ, Guchelaar H. The risk of cancer in users of statins. J Clin Oncol 2004;22:2388-94.
- Kaye JA, Jick H. Statin use and cancer risk in the General Practice Research Database. Br J Cancer 2004;90:635-7.
- Friis S, Poulsen AH, Johnsen SP, McLaughlin JK, Fryzek JP, Dalton SO, et al. Cancer risk among statin users: a population-based cohort study. Int J Cancer 2005;114:643-47.
- Jacobs EJ, Rodriguez C, Brady KA, Connell CJ, Thun MJ, Calle EE. Cholesterol-lowering drugs and colorectal cancer incidence in a large United States cohort. J Natl Cancer Inst 2006;98:69-72.
- Narisawa T, Fukaura Y, Terada K, Umezawa A, Tanida N, Yazawa K, et al. Prevention of 1,2-dimethylhydrazine-induced colon tumorigenesis by HMG-CoA reductase inhibitors, pravastatin and simvastatin, in ICR mice. Carcinogenesis 1994;15:2045-48.
- Narisawa T, Fukaura Y, Tanida N, Hasebe M, Ito M, Aizawa R. Chemopreventive efficacy of low dose of pravastatin, an HMG-CoA reductase inhibitor, on 1,2-dimethylhydrazine-induced colon carcinogenesis in ICR mice. Tohoku J Exp Med 1996;180:131-8.
- Narisawa T, Morotomi M, Fukaura Y, Hasebe M, Ito M, Aizawa R. Chemoprevention by pravastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, of N-methyl-N-nitrosourea-induced colon carcinogenesis in F344 rats. Jpn J Cancer Res 1996;87:798-804.
- Agarwal B, Rao CV, Bhendwal S, Ramey WR, Shirin H, Reddy BS, et al. Lovastatin augments sulindac-induced apoptosis in colon cancer cells and potentiates chemopreventive effect of sulindac. Gastroenterology 1999;117:838-47.
- Rao CV, Newmark HL, Reddy BS. Chemopreventive effect of farnesol and lanosterol on colon carcinogenesis. Cancer Detect Prev 2002;26:419-25.
- Kim KP, Whitehead C, Piazza G, Wargovich MJ. Combinatorial chemoprevention: efficacy of lovastatin and exisulind on the formation and progression of aberrant crypt foci. Anticancer Res 2004;24:1805-11.
- Reddy BS, Wang CX, Kong AN, Khor TO, Xi Zheng, Steele VE, et al. Prevention of azoxymethane-induced colon cancer by combination of low doses of atorvastatin, aspirin, and celecoxib in F344 rats. Cancer Res 2006;66:4542-6.
- Hirai A, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Tatsuno I, et al. Geranylgeranylated rho small GTPase(s) are essential for the degradation of p27Kip1 and facilitate the progression for G1-S phase in growth-stimulated rat FRTL-5 cells. J Biol Chem 1997;272:13-6.
- Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, et al. Inhibition of the 3-hydroxy-3 methylglutaryl-coenzyme A reductase pathway induces p53-independent transcriptional regulation of p21(WAF1/CIP1) in human prostate carcinoma cells. J Biol Chem 1998;273:10618-23.
- Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis 2001;22:1139-48.
- Wachtershauser A, Akoglu B, Stein J. HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2. Carcinogenesis 2001;22:1061-7.
- Swamy MV, Cooma I, Reddy BS, Rao CV. Lamin B, caspase-3 activity, and apoptosis induction by a combination of HMG-CoA reductase inhibitor and COX-2 inhibitors: a novel approach in developing effective chemopreventive regimens. Int J Oncol 2002;20:753-9.
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science 2001;294:1866-70.
- Sporn MB, Suh N. Chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer 2002;2:537-43.
- Shulman AI, Mangelsdorf DJ. Retinoid x receptor heterodimers in the metabolic syndrome. N Engl J Med 2005;353:604-15.
- Darwiche N, Scita G, Jones C, Rutberg S, Greenwald E, Tennenbaum T, et al. Loss of retinoic acid receptors in mouse skin and skin tumors is associated with activation of the rasHa oncogene and high risk for premalignant progression. Cancer Res 1996;56:4942-9.
- Jiang SY. Expression of nuclear retinoid receptors in normal, premalignant and malignant gastric tissues determined by in situ hybridization. Br J Cancer 1999;80:206-14.
- Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor γ (PPARγ) heterodimer. A basis for new therapeutic strategies. J Exp Med 2001;193:827-38.
- Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, et al. Adenomatous polyposis coli (APC)-independent regulation of β-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem 2003;278:29954-62.
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell 1995;83:841-50.
- Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 1990;345:224-29.
- Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong US, Evans RM. A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell 1991;66:555-61.
- Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, et al. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992;68:397-406.
- Mukherjee R, Davies PJA, Crombie DL, Bischoff ED, Cesario RM, Jow L, et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997;386:407-10.
- Bischoff ED, Gottardis MM, Moon TE, Heyman RA, Lamph WW. Beyond tamoxifen: the retinoid X receptor-selective ligand LGD1069 (Targretin) causes complete regression of mammary carcinoma. Cancer Res 1998;58:479-84.
- Wu K, Kim N-T, Rodriquez JL, Hilsenbeck SG, Mohsin SK, Xu XC, et al. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD 1069. Cancer Epidemiol Biomarkers Prev 2002;11:467-74.
- Boehm MF, Zhang L, Li Zhi, McClurg MR, Berger E. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J Med Chem 1995;38:3146-55.
- Vuligonda V, Thacher SM, Chandraratna RA. Enantioselective syntheses of potent retinoid X receptor ligands: differential biological activities of individual antipodes. J Med Chem 2001;44:2298-303.
- Deroo JB, Korach KS. Estrogen receptors and human disease. J Clin Invest 2006;116:561-70.
- Chambliss KL, Yuhanna IS, Anderson RGW, Mendelsohn ME, Shaul PW. ERβ has nongenomic action in caveolae. Mol Endocrinol 2002;16:938-46.
- Govind AP, Thampan RV. Membrane associated estrogen receptors and related proteins: Localization at the plasma membrane and the endoplasmic reticulum. Mol Cell Biochem 2003;253:233-40.
- Zivadinovic D, Watson SC. Membrane estrogen receptor-α levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res 2005;7:R130-44.
- Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol 2005;19:1951-9.
- Calle EE, Miracle-McMahill HL, Thun MJ, Health CW Jr. Estrogen replacement therapy and risk of fatal colon cancer in a prospective cohort of postmenopausal women. J Natl Cancer Inst 1995;87:517-23.
- Newcomb PA, Storer BE. Postmenopausal hormone use and risk of large-bowel cancer. J Natl Cancer Inst 1995;87:1067-71.
- Singh S, Langman MJ. Oestrogen and colonic epithelial cell growth. Gut 1995;37:737-9.
- Grodstein F, Newcomb PA, Stampfer MJ. Postmenopausal hormone therapy and the risk of colorectal cancer: A review and meta-analysis. Am J Med 1999;106:574-84.
- Olofinlade O, Grewal P, Gualtieri N. Colorectal carcinoma in young female patients. Gastroenterology 2000;118 Suppl 2:A756.
- Sturgeon SR, Schairer C, Brinton LA, Pearson T, Hoover RN. Evidence of healthy estrogen user survivor effect. Epidemiology 1995;6:227-31.
- kampman E, potter JD, slattery ML, can BJ, Edwards S. Hormone replacement therapy, reproductive history, and colon cancer: a multicenter case-control study in the United States. Cancer Causes Control 1997;8:146-58.
- Troisi R, Schairer C, Chow WH, Schatzkin A, Brinton LA, Fraumeni JF Jr. A prospective study of menopausal hormones and risk of colorectal cancer (United States). Cancer Causes Control 1997;8:130-8.
- Fernandez E, La Vecchia C, Braga C. Hormone replacement therapy and risk of colon and rectal cancer. Cancer Epidemiol Biomarkers Prev 1998;7:329-33.
- Womens Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy. The women’s Health Initiative randomized Controlled Trial. JAMA 2004;291:1701.
- Collaborative group on hormonal factors in breast cancer. Breast cancer and hormone replacement therapy:collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet 1997;350:1047-50.
- Waliszewski P, Blaszczyk M, Wolinska-Witort E, Drews M, Snochowski M, Hurst RE. Molecular study of sex steroid receptor gene expression in human colon and in colorectal carcinomas. J Surg Oncol 1997;64:3-11.
- Raigoso P, Sanz L, Vizoso F. Estrogen and progesterone receptors in colorectal cancer and surrounding mucosa. Int J Biol Markers 2001;16:262-7.
- Speer G, Cseh K, Winkler G, Takács I, Barna I, Nagy Z, et al. Oestrogen and vitamin D receptor (VDR) genotypes and the expression of ErbB-2 and EGF receptor in human rectal cancers. Eur J Cancer 2001;37:1463-8.
- Witte D, Chirala M, Younes A, Li Y, Younes M. Estrogen receptor β is expressed in human colorectal adenocarcinoma. Hum Pathol 2001;32:940-4.
- Berta L, Fronticelli Baldelli C, Fazzari A, Radice E, Bargoni A, Frairia R, et al. Sex steroid receptors, secondary bile acids and colorectal cancer. A possible mechanism of interaction. Panminerva Med 2003;45:261-6.
- Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, et al. Oestrogen receptor β (ERβ) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour’s dedifferentiation. Eur J Cancer 2003;39:1251-8.
- Javid SH, Moran AE, Carothers AM, Redston M, Bertagnolli MM. Modulation of tumor formation and intestinal cell migration by estrogens in the ApcMin/+ mouse model of colorectal cancer. Carcinogenesis 2005;26:587-95.
- Narayan S, Rajakumar G, Prouix H, Singh P. Estradiol is trophic for colon cancer in mice: Effect on omithine decarboxylase and c-myc messenger RNA. Gastroenterology 1992;103:1823-32.
- Katzenellenbogen BS, Montano MM, Ediger TR, Sun J, Ekena K, Lazennec G, et al. Estrogen receptors: selective ligands, partners, and distinctive pharmacology. Recent Prog Horm Res 2000;55:163-93.
- Katzenellenbogen BS, Choi I, Delage-Mourroux R, Ediger TR, Martini PG, Montano M, et al. Molecular mechanisms of estrogen action: selective ligands and receptor pharmacology. J Steroid Biochem Mol Biol 2000;74:279-85.
- Di Leo A, Messa C, Russo F, Misciagna G, Guerra V, Taveri R, et al. Prognostic value of cytosolic estrogen receptors in human colorectal carcinoma and surrounding mucosa. Preliminary results. Dig Dis Sci 1994;39:2038-42.
- Foley EF, Jazaeri AA, Shupnik MA, Jazaeri O, Rice LW. Selective loss of estrogen receptor beta in malignant human colon. Cancer Res 2000;60:245-8.
- Campbell-Thompson M, Lynch IJ, Bhardwaj B. Expression of estrogen receptor (ER) subtypes and ERbeta isoforms in colon cancer. Cancer Res 2001;61:632-40.
- Qiu Y, Waters CE, Lewis AE, Langman MJ, Eggo MC. Oestrogen-induced apoptosis in colonocytes expressing oestrogen receptor beta. J Endocrinol 2002;174:369-77.
- Cho NC, Javid SH, Carothers AM, Redston M, Bertagnoll MM. Estrogen receptors alpha and beta are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Res 2007;67:2366-72.
- Jassam N, Bell SM, Speirs V, Quirke P. Loss of expression of oestrogen receptor beta in colon cancer and its association with Dukes’staging. Oncol Rep 2005;14:17-21.
- Xie L, Yu J, Luo H. Expression of estrogen receptor beta in human colorectal cancer. World J Gastroenterol 2004;10:214-17.
- Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, et al. Role of estrogen receptor β in colonic epithelium. Proc Natl Acad Sci USA 2006;103:2959-64.
- Kansra S, Yamagata S, Sneade L, Foster L, Ben-Jonathan N. Differential effects of estrogen receptor antagonists on pituitary lactotroph proliferation and prolactin release. Mol Cell Endocrinol 2005;239:27-36.
- Kuruppu D, Christophi C, Bertram JF, O, Brien PE. Tamoxifen inhibits colorectal cancer metastases in the liver: A study in a murine model. J Gastroenterol Hepatol 1998;13:521-7.
- Rutqvist L, Johansson H, Signomklao T, Johansson U, Fornander T, Wilking N. Adjuvant tamoxifen therapy for early stage breast cancer and second primary malignancies. J Natl Cancer Inst 1995;87:645.
- Grodstein R, Newcomb PA, Stampfer MJ. Postmenopausal hormone therapy and the risk of colorectal cancer: A review and meta-analysis. Am J Med 1999;106:574.
- Cook L, Weiss N, Pharris-Ciurej N, Schwartz S, White E. Colorectal cancer following tamoxifen therapy for breast cancer (United States). Cancer Causes Control 2001;12:405.
- Fabian CJ, Kimler BF. Selective estrogen-receptor modulators for primary prevention of breast cancer. J Clin Oncol 2005;23:1644-55.
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effect of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006;295:2727-41.
- Ettinger B, Black DM, Mitlak BH, Knickerbocker RK, Nickelsen T, Genant HK, et al. Multiple Outcomes of Raloxifene Evaluation Investigators. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. JAMA 1999;282:637-45.
- Siris ES, Harris ST, Eastell R, Zanchetta JR, Goemaere S, Diez-Perez A, et al. Skeletal effects of raloxifene after 8 years: Results from the Continuing Outcomes Relevant to Evista (CORE) study. J Bone Miner Res 2005;20:1514-24.
- Picariello L, Fiorelli G, Martineti V, Tognarini I, Pampaloni B, Tonelli F, et al. growth response of colon cancer cell lines to selectve estrogen receptor modulators. Anticancer Res 2003;23:2419-24.
- Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, et al. Human estrogen receptor β-gene structure,chromosomal localization,and expression pattern. J Clin Endocrinol Metab 1997;82:4258-65.
- Slattery ML, Sweeney C, Murtaugh M, Ma KN, Wolff RK, Potter JD, et al. Associations between ERα, ERβ, and AR Genotypes and Colon and Rectal Cancer. Cancer Epidemiol Biomarkers Prev 2005;14:2936-42.
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997;11:3286-305.
- Huelsken J, Birchmeier W. New aspects of Wnt signaling pathways in higher vertebrates. Curr Opin Genet Dev 2001;11:547-53.
- Lee HK, Choi YS, Park YA, Jeong S. Modulation of oncogenic transcription and alternative splicing by β-catenin and an RNA aptamer in colon cancer cells. Cancer Res 2006;66:10560-6.
- Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/β-catenin/Tcf signaling axis: Wnt you like to know? Endocrine Rev 2005;26:898-15.
- Kouzmenko AP, Takeyama K-I, Ito S, Furutani T, Sawatsubashi S, Maki A. Wnt/β-catenin and estrogen signaling converge in vivo. J Biol Chem 2004;279:40255-8.
- Powell SM, Zilk N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235-7.
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996;272:1023-6.
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996;382:638-42.
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S. Axin, a negative regulator of the Wnt signalling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J 1998;17:1371-84.
- Sakanaka C, Weiss JB, Williams LT. Bridging of β-catenin and glycogen synthase kinase-3β by axin and inhibition of β-catenin-mediated transcription. Proc Natl Acad Sci USA 1998;95:3020-3.
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK-3β. Science 1998;280:596-9.
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-Catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130-4.
- Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996;382:638-42.
- Hugh TJ, Dillon SA, O’ Dowd G, Getty B, Pignatelli M, Poston GJ, et al. Beta-catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer 1999;82:504-11.
- Wong NA, Pignatelli M. β-catenin – A linchpin in colorectal carcinogenesis? Am J Pathol 2002;160:389-401.
- Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates β-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem 2006;281:17751-7.
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275:1787-90.
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997;275:1784-7.
- Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W, et al. Wnt/B-catenin signaling pathway as novel cancer drug targets. Curr Cancer Drug Targets 2004;4:653-71.
- Dihlmann S, von Knebel Doeberitz M. Wnt/β-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int J Cancer 2005;113:515-24.
- de Vogel S, van Engeland M, Luchtenborg M, de Bruine AP, Roemen GM, Lentjes MH, et al. Dietary folate and APC mutations in sporadic colorectal cancer. J Nutr 2006;136:3015-21.
- Cole F, Baron JA, Sandler RS, Haile RW, Ahnen DJ, Bresalier RS, et al. Folic acid for the prevention of colorectal adenomas. JAMA 2007;297:2351-9.
- Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits β-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear β-catenin-TCF association. Proc Natl Acad Sci USA 2003;100:12584-9.
- Dean NM, Bennett CF. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003;22:9087-96.
- Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, et al. Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell 2004;5:91-102.
- Prolexys Pharmaceuticals[homepage on the Internet]. Small Molecule Inhibitors of the Beta-catenin Pathway. [updated 2007 Dec 1; cited 2007 Dec 20] Available from: .http://www.prolexys.com/public/Prolexys_Beta-Catenin.pdf
- Borgeat P, Hamberg M, Samuelsson B. Transformation of arachidonic acid and homo-gamma-linolenic acid by rabbit polymorphonuclear leukocytes. Monohydroxy acids from novel lipoxygenases. J Biol Chem 1976;251:7816-20.
- Ye YN, Liu ESL, Shin VY, Cho CH. Involvement of 5-lipoxygenase, matrix metalloproteinase-2 and vascular endothelial growth factor in the colonic tumorigenesis promoted by passive cigarette smoking. Am Assoc Cancer Res Proc 2003;44:95.
- Ye YN, Liu ES, Shin VY, Wu WK, Luo JC, Cho CH. Nicotine promoted colon cancer growth via epidermal growth factor receptor, c-Src, and 5-lipoxygenase-mediated signal pathway. J Pharmacol Exp Ther 2004;308:66-72.
- Ye YN, Wu WKK, Shin VY, Cho CH. A mechanistic study of colon cancer growth promoted by cigarette smoke extract. Eur J Pharmacol 2005;519:52-7.
- Ikawa H, Kamitani H, Calvo BF, Foley JF, Eling TE. Expression of 15-lipoxygenase-1 in human colorectal cancer. Cancer Res 1999;59:360-6.
- Ohd JF, Nielsen CK, Campbell J, Landberg G, Lofberg H, Sjolander A. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectaladenocarcinomas. Gastroenterology 2003;124:57-70.
- Kennedy TJ, Talamonti M, Ujiki M, Ding XZ, Ternent CA, Bell RH Jr, et al. Lipoxygenase expression in colon polyps and inhibition of colon cancer growth by lipoxygenase blockade. J Am Coll Surg 2004;199:78.
- Soumaoro LT, Iida S, Uetake H, Ishiguro M, Takagi Y, Higuchi T, et al. Expression of 5-Lipoxygenase in human colorectal cancer. Gastroenterology 2006;12:6355-60.
- Öhd JF, Wikström K, Sjölander A. Leukotrienes induce cell-survival signaling in intestinal epithelial cells. Gastroenterology 2000;119:1007-18.
- Dahlen SE, Haeggstrom JZ, Samuelsson B, Rabe KF, Leff AR. Leukotrienes as targets for treatment of asthma and other diseases. Current basic and clinical research. Am J Respir Crit Care Med 2000;161:S1.
- Chen X, Wang S, Wu N, Rao SM, Buchler MW, Friess H. Overexpression of 5-lipoxygenase in rat and human esophageal adenocarcinoma and inhibitory effects of zileuton and celecoxib on carcinogenesis. Clin Cancer Res 2004;10:6703-09.
- Hennig R, Grippo P, Ding XZ, Rao SM, Buchler MW, Friess H, et al. 5-lipoxygenase, a marker for early pancreatic pancreatic intraepithelial neoplastic lesions. Cancer Res 2005;65:6011-6.
- Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Se CN. selectivity of recombinant human leukotrienes D4, leukotrienes B4 and lipoxins A4 receptors with aspirin-triggered 15 epi-LXA4 and regulation of vascular and inflammatory responses. Am J Pathol 2001;158:3-9.
- Emerson MR, LeVine SM. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res 2004;17:140-5.
- Groser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest 2006;116:4-15.
- Goossens L, Pommery N, Henichart JP. COX-2/5-LOX dual acting anti-inflammatory drugs in cancer chemotherapy. Curr Topics Med Chem 2007;7:283-96.
- Leval X, Julemont F, Delarge J, Pirotte B, Dogne JM. New trends in dual 5-LOX/COX inhibition. Curr Med Chem 2002;9:941-62.
- Horizoe T, Nagakura N, Chiba K, Shirota H, Shinoda M, Numata H, et al. Effects of ER-34122, a novel dual 5-lipoxygenase/cyclooxygenase inhibitor, on indices of early articular lesion in MRL/MpJ-lpr/lpr mice. Inflamm Res 1999;48:432-6.
- Martel-Pelletier J, Lajeunesse D, Reboul P, Pelletier JP. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann Rheum Dis 2003;62:501-9.
- Kirchner T, Aparicio B, Argentieri DC, Lau CY, Ritchie DM. Effects of tepoxalin, a dual inhibitor of cyclooxygenase/5-lipoxygenase, on events associated with NSAID-induced gastrointestinal inflammation. Prostaglandins Leukot Essent Fatty Acids 1997;56:417-23.
- Waldman SA, Vitow C, Osborne B, Gillen L, Argentieri DC, Wong FA, et al. Pharmacokinetics and pharmacodynamics of tepoxalin after single oral dose administration to healthy volunteers. J Clin Pharmacol 1996;36:462-8.
- Depre M, Van Hecken A, Verbesselt R, Verpooten GA, Arnout J, Brunner F, et al. Biochemical activity, pharmacokinetics and tolerability of tepoxalin, a cyclooxygenase/5-lipoxygenase inhibitor, in man. Int J Clin Pharmacol Res 1996;16:1-8.
- Klesser B, Bias P, Buchner A, Elsaesser R. Licofelone (ML3000), an inhibitor of COX-1, COX-2 and 5-LOX, has little or no effect on the gastric mucosa after 4 weeks of treatment. Ann Rheum Dis 2002;61:130.
- Alvaro-Gracia JM. Licofelone—clinical update on a novel LOX/COX inhibitor for the treatment of osteoarthritis. Rheumatology 2004;43:i21-5.
- Levy DE, Lee CK. What does Stat3 do? J Clin Invest 2002;109:1143-8.
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000;19:2474-88.
- Ma XT, Wang S, Ye YJ, Du RY, Cui ZR, Somsouk M. Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J Gastroenterol 2004;10:1569-73.
- Christine R, Sylvie R, Erik B, Geneviève P, Amélie R, Gérard R, et al. Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3) – and vascular endothelial growth factor–mediated cellular invasion and tumor growthh. Cancer Res 2005;65:195-202.
- Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005;7:545-55.
- Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol 2005;167:969-80.
- Bromberg J. Stat proteins and oncogenesis. J Clin Invest 2002;109:1139-42.
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell 1999;98:295-303.
- Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 2001;107:351-62.
- Epling-Burnette PK, Zhong B, Bai F, Jiang K, Bailey RD, Garcia R, et al. Cooperative regulation of Mcl-1 by Janus kinase/Stat and phosphatidylinositol 3-kinase contribute to granulocyte–macrophage colony-stimulating factor–delayed apoptosis in human neutrophils. J Immunol 2001;166:7486-95.
- Shen Y, Devgan G, Darnell JE Jr, Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci USA 2001;98:1543-8.
- Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive STAT3 activity upregulates VEGF expression and tumor angiogenesis. Oncogene 2002;21:2000-8.
- Wei WD, Le X, Zheng L, Wang L, Frey JA, Gao AC. STAT3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastatis. Oncogene 2003;22:319-29.
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE Jr. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol 1998;18:2553-8.
- Turkson J, Bowman T, Garcia R, Caldenhoven E, Rolf P, De Groot RP, et al. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol 1998;18:2545-52.
- Bertrand JR, Pottier M, Vekris A, Opolon P, Maksimenko A, Malvy C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem Biophys Res Commun 2002;296:1000-4.
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002;2:243-7.
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins HI, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 2004;10:816-20.
- Lv W, Zhang C, Hao J. RNAi technology: A revolutionary tool for the colorectal cancer therapeutics. World J Gastroenterol 2006;12:4636-9.
- Huang ZG, Ran ZH, Lu W, Xiao SD. Analysis of gene expression profile in colon cancer using the Cancer Genome Anatomy Project and RNA interference. Chin J Dig Dis 2006;7:97-102.
- George S C, Bharati B. Cyclooxygenase-2 knockdown by RNA interference in colon cancer. Int J Oncol 2006;28:543-9.
- Jiang M, Milner J. Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev 2003;17:832-7.
- Yang M, Zhong WW, Srivastava N, Slavin A, Yang J, Hoey T, et al. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the beta-catenin pathway. Proc Natl Acad Sci USA 2005;102:6027-32.
- Hammond SM, Caudy AA, Hannon GJ. Post-transcriptional gene silencing by double-stranded RNA. Nat Rev Genet 2001;2:110-9.
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan micro-RNAs. Nat Rev Genet 2004;5:396-400.
- Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev 2005;15:563-8.
- Lim LP, Lau NC, Garrett-engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005;433:769-73.
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15-20.
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003;115:787-98.
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol 2004;2:e363.
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev 2004;18:1165-78.
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet 2005;37:495-500.
- Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005;65:7065-70.
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005;65:9628-32.
- Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LAJ, Sjoblom T, et al. The colorectal microRNAome. Proc Natl Acad Sci USA 2006;103:3687-92.
- Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N, et al. Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer 2006;5:29.
- Calin GA, Croce CM. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res 2006;66:7390-4.
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006;6:857-66.
- Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 2006;6:259-69.
- Hammond SM. MicroRNAs as oncogenes. Curr Opin Genet Dev 2006;16:4-9.
- Chang T-C, Wentzel EA, Kent AO, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007;26:745-52.
- Michael MZ, O’Connor SM, Van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 2003;1:882-91.
- Akao Y, Nakagawa Y, Naoe T. MicroRNA-143 and -145 in colon cancer. DNA Cell Biol 2007;26:311-20.
- Qiao D, Zeeman AM, Deng W, Looijenga LH, Lin H. Molecular characterization of hiwi, a human member of the piwi gene family whose overexpression is correlated to seminomas. Oncogene 2002;21:3988-99.
- Liu X, Sun Y, Guo J, Ma H, Li J, Dong B, et al. Expression of hiwi gene in human gastric cancer was associated with proliferation of cancer cells. Int J Cancer 2006;118:1922-9.