
Pharmacokinetic characteristics of L-valyl-ara-C and its implication on the oral delivery of ara-C1
Introduction
The clinical utility of ara-C, one of the most effective drugs against the acute myeloid leukemia, acute lymphoblastic leukemia and other hematological malignancies[1,2], is severely limited by rapid deamination primarily in the liver, spleen and gastrointestinal mucosa[3,4]. Consequently, ara-C has a very short plasma half-life as well as low systemic exposure, and must be administered in continuous infusion or on a complex schedule to provide maximum therapeutic efficacy[3,4]. Therefore, considerable efforts have been directed at an enhancement of the therapeutic efficacy of ara-C, and many prodrug strategies have been explored for the oral delivery of ara-C with varied degrees of success[5–8]. However, few have led to an approved product and thus, there continues to be a great need for improved methods to deliver pharmacologically active ara-C.
Recently, our group synthesized the L-valyl-ara-C by masking the N4-amino group of the cytosine ring in ara-C with L-valine in order to enhance the intestinal absorption of ara-C, and evaluated the cellular uptake characteristics of L-valyl-ara-C in Caco-2 cells[9]. Compared to ara-C, L-valyl-ara-C appeared to be stable in the intestinal lumen and exhibited 5 fold higher cellular uptake via the carrier-mediated transport pathways. Those results indicated that L-valyl-ara-C could be a promising candidate for the oral delivery of ara-C. Therefore, the present study aimed to examine the pharmacokinetic characteristics of L-valyl-ara-C in rats and evaluate its utility as a potential oral delivery system of ara-C in vivo.
Materials and methods
Materials Ara-C, acyclovir and 5-bromo-2'-deoxyuridine were purchased from Sigma Chemical Co (St Louis, MO, USA). Acetonitrile and methanol were obtained from Merck Co (Darmstadt, Germany). All other chemicals were of reagent grade and all solvents were of HPLC grade. AML2 and L1210 cells were purchased from Korean Cell Line Bank (Seoul, Korea).
Cells The L1210 cells were routinely maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum and penicillin (100 U/mL)/streptomycin (50 mg/mL). The AML-2 cells were grown in DMEM supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (50 mg/mL). All of the cells were maintained in an atmosphere of 5% CO2 and 90% relative humidity at 37 ºC.
Synthesis of L-valyl-ara-C Prodrug was synthesized as previously described by Cheon et al[9], and identified by the 1H-NMR, 13C-NMR and EI-MS spectrometers.
In vitro stability study Stabilities of L-valyl-ara-C were evaluated at 37 °C by incubating a drug solution (10 µmol/L) in the fresh plasma and cell homogenates. Enzymatic cleavage of amide bond between valine and ara-C was also measured in the presence of plasmin. Plasmin 30 µL (10 Cu/mL) was added to the thermostated buffer composed of 50 mmol/L Tris-HCl, pH 7.4 and 110 mmol/L NaCl, followed by the addition of the drug solution (10 µmol/L) to a final volume of 1.0 mL at 37 °C. At each time point, the metabolic reaction was stopped by adding ice-cold acetonitrile followed by vigorous mixing. The mixture was then centrifuged at 3000 r/min for 10 min at 4 °C and the supernatant was filtered through a membrane filter (0.45 μm) and analyzed by HPLC. The chemical stability of L-valyl-ara-C was also examined in aqueous solutions of different pH (2.0, 7.4 and 10).
Antiproliferative assays Cells were resuspended at a density of 1×105 cells/mL in growth medium containing serial dilutions of test drugs. Cell viability was determined after 96 h at 37 °C by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method, as previously described by Pauwels et al[10]. Cell growth at each drug solution was expressed as percentage of untreated controls, and IC50 was determined as the concentration resulting in 50% growth inhibition.
Pharmacokinetic studies in rats Male Sprague-Dawley rats (270–300 g) were purchased from Dae Han Laboratory Animal Research and Co (Choongbuk, Korea), and given a normal standard chow diet (N
HPLC assay Drug concentrations were determined by a HPLC assay described as follows: acyclovir and 5-bromo-2'-deoxyuridine were used as the internal standard for the assay of ara-C and L-valyl-ara-C, respectively. The chromatographic system, consisting of a pump (LC-10AD), an automatic injector (SIL-10A) and a UV detector (SPD-10A; Shimadzu Scientific Instruments, Japan), was set at 272 nm. An octadecylsilane column (Gemini C18, 4.6 mm×250 mm, 5 µm; Phenomenex, Torrance, CA, USA) was eluted with a mobile phase at a flow rate of 1.0 mL/min. The mobile phase consisted of 0.01 mol/L ammonium acetate buffer (pH 6.5):acetonitrile (93:7, v/v) for L-valyl-ara-C and 0.01 mol/L ammonium acetate buffer (pH 4.5):acetonitrile (99:1, v/v) for ara-C. The calibration curve from the standard samples was linear over the concentration range of 0.01 µg/mL to 5 µg/mL. The limit of detection was 0.01 µg/mL.
Pharmacokinetic analysis Non-compartmental pharmacokinetic analysis was performed using WinNonlin® (Pharsight Corp, CA, USA). The area under the plasma concentration-time curve (AUC) was calculated using the linear trapezoidal method. The maximum plasma concentration (Cmax) and the time to reach the maximum plasma concentration (Tmax) were observed values from the experimental data. The terminal elimination rate constant (λz) was estimated from the slope of the terminal phase of the log plasma concentration-time points fitted by the method of least-squares, and then the terminal elimination half-life (T1/2) was calculated as 0.693/λz. Additional estimated parameters using noncompartmental pharmacokinetic analysis were systemic plasma clearance (CL) and the volume of distribution (Vdss).
Statistical analysis All the means are presented with their standard deviation. An unpaired Student’s t-test was used to determine the significant difference between treat-ments. A P value <0.05 was considered statistically signi-ficant.
Results
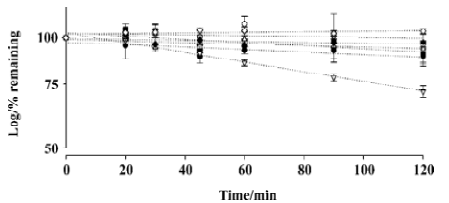
In vitro metabolic stability studies The kinetics of L-valyl-ara-C degradation were studied in various conditions. As shown in Figure 1, L-valyl-ara-C appeared to be stable in fresh rat plasma and leukemia cell homogenates. The degradation of L-valyl-ara-C was negligible in fresh rat plasma and leukemia cell homogenates over the 2 h incubation period. In addition, the metabolic stability of L-valyl-ara-C was assessed using pure serine protease plasmin, and the results indicated that L-valyl-ara-C was resistant to the degradation by plasmin. The chemical stability of L-valyl-ara-C was also examined in aqueous solutions of different pH (2.0, 7.4, and 10). While L-valyl-ara-C was stable in aqueous solutions of pH 7.4 and pH 10, the hydrolysis of amide bond appeared to be rather accelerated in acidic pH and the disappearance half-life of L-valyl-ara-C was approximately 4 h in the aqueous solution of pH 2.0 (Figure 1).
Antiproliferative assays The antiproliferative activity of ara-C and L-valyl-ara-C was evaluated against leukemia cells such as AML-2 and L1210. The IC50 values were 3.89±1.11 and 606.0±87.3 µmol/L for ara-C and L-valyl-ara-C, respectively in the AML-2 cells. In the case of the L1210 cells, the IC50 values were 0.25±0.03 and 37.5±6.69 µmol/L for ara-C and L-valyl-ara-C, respectively. In both types of cells, the antiproliferative activity of L-valyl-ara-C was approximately 150 fold less potent than ara-C.
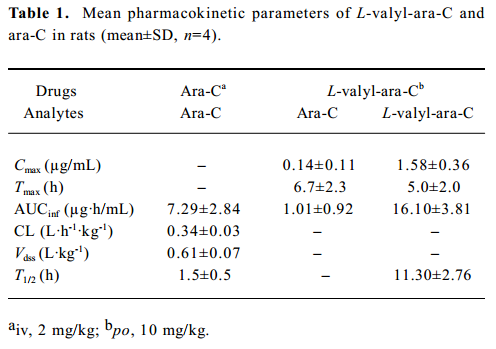
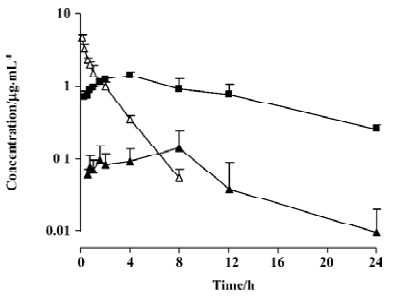
Pharmacokinetic studies The plasma pharmacokinetic profiles of L-valyl-ara-C and ara-C were determined in rats and summarized in Table 1 and Figure 2. After an oral administration of L-valyl-ara-C, the release of ara-C from the prodrug was observed in the plasma although the systemic exposure of the prodrug was much higher than that of ara-C. The bioavailability of ara-C was about 4% via the prodrug administration in the rats.
Full table
Discussion
The development of oral alternatives to intravenous administration of ara-C would not only avert the high costs dictated by hospital treatments, but also be more patient friendly. In addition, it may also be possible to incorporate prodrug strategies that could reduce undesirable side effects such as toxicity, and improve therapeutic action into such oral alternatives. In our previous studies, L-valyl-ara-C appeared to be beneficial in enhancing the cellular uptake of ara-C in Caco-2 cells[9]. However, in the present study, prodrug itself exhibited far less antiproliferative activity than ara-C in AML-2 and L1210 cells and thus, the rate of conversion of the prodrug to the pharmacologically-active parent drug after membrane transport would determine the therapeutic effectiveness of L-valyl-ara-C.
If the prodrug is stable in plasma and hydrolyzed mainly near the target cells, this should result in a continuous and relatively high concentration of the active agent around target cells. Many types of malignant cells and human tumors display increased concentrations of the protease plasminogen activators that convert plasminogen to the highly-active protease, plasmin[11–13]. Leukemic cells also secrete these enzymes[14]. Since plasmin rapidly cleaves various low molecular weight compounds coupled to appropriate peptide specifiers, the coupling of such peptide specifiers to anticancer drugs might create ‘prodrugs’ which would be locally activated by tumor-associated plasmin and consequently would be less toxic to normal cells. For example, Carl et al[15] reported that the peptide prodrugs of several anticancer agents designed to be specific plasmin substrates showed selective cytotoxicity. Since L-valyl-ara-C is a pepti-domimetic prodrug and appears to be stable in plasma or leukemia cell homogenates, the potential bioconversion of L-valyl-ara-C to ara-C in the surroundings of the leukemia cells was examined using pure plasmin. As shown in Figure 1, L-valyl-ara-C exhibited high resistance to the degradation by plasmin, implying that L-valyl-ara-C may not be a substrate of plasmin. Overall, in vitro stability studies have indicated that L-valyl-ara-C is metabolically stable in plasma and leukemia cells. However, in the case of chemical stability, the hydrolysis of prodrug appeared to be rather accelerated in acidic pH. Considering that the tumor pH is on average lower than the pH of normal tissues[16], chemical hydrolysis of L-valyl-ara-C may be more favorable in tumor cells.
As summarized in Table 1, following an intravenous administration of ara-C to rats, ara-C showed a short plasma half-life of 1.5 h, and its volume of distribution was greater than total body water in the rats[17]. The pharmacokinetic parameters of ara-C obtained from the present study appeared to be comparable to those from previous studies[18–20]. Following an oral administration of L-valyl-ara-C to the rats, the appearance of ara-C was observed in the plasma, implying that the prodrug conversion to the parent could occur by intestinal or hepatic metabolism (Figure 2). L-valyl-ara-C is a peptidyl derivative of ara-C and thus, during intestinal absorption, it may interact with peptidases which catalyze the hydrolysis of peptide bonds in peptidyl derivatives or the hydrolysis of various simple amides[21]. However, as implicated by the results from the in vitro stability study of L-valyl-ara-C, the metabolic conversion of L-valyl-ara-C appeared to be minimal in the rats. Consequently, the systemic exposure (AUC) of the prodrug was much higher than that of ara-C, and the bioavailability of ara-C was low (about 4%) via the prodrug administration. Therefore, the metabolic conversion of the prodrug to the parent did not appear to be sufficient to ensure therapeutic effectiveness in the treatment of tumors, although L-valyl-ara-C could prevent the rapid deamination of ara-C by masking the N4-amino group of the cytosine ring. Further studies should be required for the considerable tuning of the metabolic stability of prodrugs by varying its amino acid component.
In conclusion, the amide bond of L-valyl-ara-C was stable against the enzymatic hydrolysis, and the utility of L-valyl-ara-C as an oral delivery system of ara-C appeared to be limited by its low metabolic conversion to ara-C in the rats.
Acknowledgement
The authors wish to express their appreciation to Mr Ming-ji JIN for his help with the animal experiment.
References
- Bryan JH, Henderson ES, Leventhal BG. Cytosine arabinoside and 6-thioguanine inrefractory acute lymphocytic leukemia. Cancer 1974;33:539-44.
- Grant S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res 1998;72:197-233.
- Ho DH, Frei E 3rd. Clinical pharmacology of 1-beta-d-arabinofuranosyl cytosine. Clin Pharmacol Ther 1971;12:944-54.
- Rustum YM, Raymakers RA. 1-Beta-arabinofuranosylcytosine in therapy of leukemia: preclinical and clinical overview. Pharmacol Ther 1992;56:307-21.
- Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev 2003;55:217-50.
- Hadfield AF, Sartorelli AC. The pharmacology of prodrugs of 5-fluorouracil and 1-beta-D-arabinofuranosylcytosine. Adv Pharmacol Chemother 1984;20:21-67.
- Wipf P, Li W. Prodrugs of ara-C. Drugs Future 1994;19:49-54.
- Wipf P, Li W, Adeyeye CM, Rusnak JM, Lazo JS. Synthesis of chemoreversible prodrugs of ara-C with variable time-release profiles. Biological evaluation of their apoptotic activity. Bioorg Med Chem 1996;4:1585-96.
- Cheon EP, Hong JH, Han HK. Enhanced cellular uptake of ara-C via a peptidomimetic prodrug, L-valyl-ara-C in Caco-2 cells. J Pharm Pharmacol 2006;58:927-32.
- Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, et al. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 1988;20:309-21.
- Axelrod JH, Reich R, Miskin R. Expression of human recombinant plasminogen activators enhances invasion and experimental metastasis of H-ras-transformed NIH 3T3 cells. Mol Cell Biol 1989;9:2133-41.
- Nagy B, Ban J, Brdar B. Fibrinolysis associated with human neoplasia: production of plasminogen activator by human tumours. Int J Cancer 1977;19:614-20.
- Tucker WS, Kirsch WM, Martinez-Hernandez A, Fink LM. In vitro plasminogen activator activity in human brain tumors. Cancer Res 1978;38:297-302.
- Wilson EL, Jacobs P, Dowdle EB. The secretion of plasminogen activators by human myeloid leukemic cells in vitro. Blood 1983;61:568-74.
- Carl PL, Chakravarty PK, Katzenellenbogen JA, Weber MJ. Protease-activated “prodrugs” for cancer chemotherapy. Proc Natl Acad Sci USA 1980;77:2224-8.
- Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res 1996;56:1194-8.
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993;10:1093-5.
- Groothuis DR, Benalcazar H, Allen CV, Wise RM, Dills C, Dobrescu C, et al. Comparison of cytosine arabinoside delivery to rat brain by intravenous, intrathecal, intraventricular and intraparenc-hymal routes of administration. Brain Res 2000;856:281-90.
- Scott-Moncrieff JC, Chan TC, Samuels ML, Cook JR, Coppoc GL, DeNicola DB, et al. Plasma and cerebrospinal fluid pharmacokinetics of cytosine arabinoside in dogs. Cancer Chemother Pharmacol 1991;29:13-8.
- Zimmerman CL. The disposition of cytosine arabinoside and its metabolite after single doses to rabbits. Biopharm Drug Dispos 1990;11:121-9.
- Bai JPF, Amidon GL. Structural specificity of mucosal cell transport and metabolism of peptide drugs: implication for oral peptide drug delivery. Pharm Res 1992;9:969-78.