
Improvement of chronic heart failure by dexamethasone is not associated with downregulation of leptin in rats1
Introduction
Leptin, a product of the OB-gene, is originally associated with energy expenditure and weight loss. The expression of leptin and its receptors has been detected in several non-adipose tissues, including the myocardium[1]. An increase in leptin has been implicated in the development of cardiovascular diseases and conditions, including coronary heart diseases[2], stenting[3], and hypertension[4]. Hyperleptinemia is involved in the increased activity of the sympathetic nerve system and is closely linked with the occurrence of cardiovascular events such as myocardial infarction[5] and stroke, suggesting that leptin may participate in proatherogenic mechanisms at vascular intima. Leptin receptors (OB-Rb or OB-RL) regulate the central actions of leptin and has also been detected in various other tissues including cardiomyocytes[1].
There is considerable interstitial fibrosis in chronic heart failure (CHF), which stiffens ventricles and impedes both contraction and relaxation. The increased expression of a number of extracellular matrix proteins, including several forms of collagen and fibronectin, matrix metalloproteinases (MMP), and the downregulation of their inhibitors [tissue inhibitor of metalloproteinases (TIMP)] are responsible for the formation of the extracellular matrix (ECM), which is intimately involved in the remodeling of the cardiac matrix[6]. Changes in MMP and TIMP expression or activity are related to an excessive reactive oxygen species (ROS) during the progression from compensated to decompensated heart failure. The state of oxidative stress associates the myocardium with the induction of cardiac remodeling, cardiomyocyte hypertrophy, the activation of MMP, and inflammatory cell infiltration[7]. Recent studies provide evidence that patients with CHF exhibit an average increase in serum levels of leptin as compared to healthy controls[5]. Leptin enhances the MMP-2 and MMP-9 expression in vitro, and increases the generation of intracellular ROS in cardiomyocytes[8–10]. Moreover, pro-inflammatory cytokines correlate well with circulating levels of leptin, and hyperleptinemia in several chronic diseases has been linked to the activation of an inflammatory marker of tumor necrosis factor (TNF-α)[11,12]. An increase in TNF-α in the plasma in clinical findings is directly correlated to the progression of CHF.
Interestingly, with anti-inflammatory and immunosuppressive properties, dexamethasone (Dex) inhibits the leptin–activated transcription (STAT) 3 signaling pathway in a cultured cell line as well as in the rat hypothalamus[13]. Dex also has protective effects against TNF-α-mediated cell death[14]; however, Dex significantly upregulates leptin levels and leptin receptors, then causes an increase in OB gene expression and leptin secretion in isolated adipose tissue from rodents and humans[15,16]. In general, leptin is a risk factor for cardiovascular diseases, and marked leptin increase in the plasma and myocardium has been found in patients of CHF. The behavior of leptin in relation to the cardiovascular system varied[17,18]; leptin might do insult to the myocardium, but it is also likely to be beneficial to the heart. It would be interesting to see whether the upregulation of leptin is an important marker only in the development of CHF[5]. Dex might inhibit the leptin pathway, and also up-regulate expression of leptin and its receptors. Thus, we hypothesized that an up-regulation of leptin levels, protein expression and its receptor OB-RL contributed to CHF, it is true, however, with Dex intervention, an up-regulation of leptin and its receptor OB-Rb might not be down-regulated despite a significant relief of CHF is achieved. It is interesting to investigate whether an elevated leptin in serum is critical to induce cardiac insufficiencyby its direct action on its receptors OB-Rb in myocardium, or it is a marker only and exerts a combined effect with other cytokines responsible to compromised cardiac function. Thus it is interesting to investigate if we could separate leptin from other inflammatory factors for its adverse relationship to CHF under certain condition. Based on the effectiveness of Dex on cardiac ischemia and heart failure, we applied Dex to observe whether hyperleptinemia and an up-regulation of leptin receptors OB-Rb could not be substantially related to CHF and Dex-induced relief of CHF was associated with suppression of TNF-α, MMP-2 and MMP-9 and ROS, when hyperleptinemia, but not with up-regulation of leptin and its receptors in myocardium.
Materials and methods
Animals Male Sprague-Dawley rats (200–220 g, aged 10 weeks) were used for the experiment. They were housed in a controlled environment and allowed free access to tap water and food.
Experimental heart failure CHF was developed by coronary artery ligation in rats for 5 weeks. Briefly, the left coronary artery was ligated between the left atrial appendage and the right ventricular outflow tract with a 6.0 silk suture. The chest was then closed in layers and air was evacuated from the chest cavity by slight lateral pressure of the thorax. Using this method, the survival rate was 60%–70% at 24 h after the operation. Sham operations were performed to open the pericardium only, but no ligation was made around the coronary artery. The rats were weighed every week and their food intake was measured to adjust the doses of Dex.
Experiment protocol The rats were divided into 3 groups: (i) the sham operation (sham) group; (ii) chronic heart failure (CHF) group; and (iii) the CHF rats treated with (Dex), which was added in the drinking water (1 µg/mL, Xianju Pharmaceutical Co Ltd, China). The actual dose of Dex was approximately 50 mg⋅kg-1⋅d-1. Water containing Dex was prepared freshly everyday. In each group, the number of animals was 10, except in the expression experiment where n=4.
Hemodynamic changes In brief, the rats were anesthetized with urethane (1.5 g/kg, ip) 6 weeks after the surgical operation, and the right carotid artery was cannulated with a micromanometer-tipped catheter (PE 50, ID 0.58 mm, OD 0.965 mm, Becton Dickinson and Company, San Jose, CA, USA) which was connected to a pressure transducer (MPA-V, the Second Military Medical University, Shanghai, China) and advanced into the left ventricle (LV). The LV (+dp/dtmax and –dp/dtmin), left systolic pressure (LVSP), and left ventricular end diastolic pressure (LVEDP) were recorded. The heart rate was monitored by lead electrocardiogram.
Cardiac morphological assay After the experiments, the LV myocardium was fixed in 10% formalin. Three cross sections in the noninfarcted myocardium, from the apex to the base, were obtained and compared among the 3 groups. Assessment of myocardial hypertrophy and interstitial fibrosis were conducted in slices with HE stain, and Masson’s trichrome stain, separately. The diameter of the cardiomyocytes was evaluated by direct measurements at ×400 magnification in cross sections that included a nuclear profile. A total of 40 cells per section were evaluated. For the evaluation of fibrosis in the myocardium, imaging scanning technique of the Masson’s stained slices was performed in 3 sections per animal and 20 fields per section, and computerized with a digital image analyzer (Image-pro Plus, Media Cybernetics Inc, Silver Spring, MD, USA). The volume of collagen fraction was calculated as the sum of all connective tissue areas divided by the total area of the image[19].
Biochemical parameters of CHF The measurement of glutamic oxaloacetate transaminase (GOT), glutamic pyruvate transaminase (GPT), malondialdehyde (MDA), superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), catalase (CAT), xanthine oxidase (XOD), lactate dehydrogenase (LDH), creatine phosphokinase (CPK) in the serum, and hydroxyproline (HYP) in the left ventricles were conducted in all the rats of the 3 groups.
Radioimmunoassays of plasma leptin Plasma leptin concentrations were measured in duplicate using a specific rat leptin radioimmunoassay kit (Linco Research, St Charles, MO, USA). The interassay coefficient of the variation was less than 6%, and the detection limit was 0.5 ng/mL.
RT-PCR Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Five micrograms of RNA was used to synthesize the first strand of cDNA using SUPERSCRIPT II RNase H-Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol, and was used as a template in the following PCR reactions.
To elucidate the mRNA expression of MMP-2, MMP-9, TIMP-1, TIMP-2 and OB-Rb (leptin receptors), RT-PCR was carried out using sense primers and antisense primers. Sense: 5'-CCCAGAAAAGATTGACGC-3' and antisense: 5'-CGACAGCATCCAGGTTAT-3' for MMP-2; sense: 5'-CGT-GGCCTAGTGACCTATG-3' and antisense: 5'-GGATAGCTC-GGTGGTGTCCT-3' for MMP-9; sense: 5'-GCTGCGGTTCT-GGGATT-3' and antisense: 5'-CCTCTGGCATCCTCTTGTT-3' for TIMP-1; sense: 5'-GAAGAAAGGAGGTTGCAGT-3' and antisense: 5'-TCCAGGAAGGGATGTCAAAG-3' for TIMP-2; sense: 5'-GCTGAGAGCACCCAGGGAACC-3' and anti-sense: 5'-GTTTCCTGGCGATGCACTGGC-3' for OB-Rb. The products were resolved on 3% agarose gel followed by ethi-dium bromide staining.
Zymography of MMP-2 and MMP-9 The proteins were extracted from the cardiac tissue. Briefly, the LV were homogenized in lysis buffer containing 1% SDS, 1 µmol/L phenylmethylsulfonyl fluoride (PMSF), and 10 µg/mL leupeptin in 50 mmol/L Tris buffer at pH 7.6. Insoluble matter was removed by centrifugation at 10 000×g for 10 min. The total protein concentration for each sample was determined using the Bradford method. Gelatin was incorporated into 10% SDS-PAGE to a final concentration of 1 mg/mL. After electrophoresis, the proteins in the gel were denatured by incubation for 30 min (2×15 min) in 2.5% Triton X-100. The gels were subsequently incubated overnight at 37 °C in 50 mmol/L Tris-HCl, pH 7.4, containing 10 mmol/L of calcium chloride. Bands of lytic activity were visualized as zones of clearing after staining with Coomassie brilliant blue G-250. To verify MMP activity, identical gels were incubated overnight in the presence of 20 mmol/L of EDTA, an inhibitor of MMP, and 2 mmol/L of PMSF, a serine protease inhibitor.
Western blotting For the quantitative analysis of the leptin protein level in the myocardium, the LV tissue (100–200 mg) was homogenized in 4 volumes of extraction buffer and centrifuged at 10 000×g for 10 min. After determination of the protein concentration, the supernatants were stored at –20 °C until use. An aliquot was heated to 95 °C, and size-fractionated on 10% SDS-PAGE. The extracted protein was transferred to a nitrocellulose membrane and blocked with nonfat milk (5% w/v), followed by incubation with first antibody (1:100, BA1231, Boster Biological Technology Ltd, Wuhan, China) for another 1 h. After 3 washes, the blot was incubated with horseradish peroxidase conjugated goat secondary antibody IgG (1:1000) for an additional 1 h. Antigen was detected with a DAB kit. A linear relationship between the density of blots and the protein load was observed when 20, 40, 60, 80, and 100 μg of membrane protein was used per lane.
Statistic analysis GraphPad Prism, version 4.0 (GraphPad Software Inc, San Diego, CA, USA), was used to analyze the results. Data are presented as mean±SD. The paired Student’s t-test was used for the statistical comparison of mean values between 2 experimental groups. One-way ANOVA and Bonferroni test were used to compare mean values between all experimental groups. A value of P<0.05 was considered statistically significant.
Results
Improvement of hemodynamics and cardiac remodeling Chronic coronary ligation markedly deteriorated cardiac systolic LVSP (-18.4%) and LV +dp/dtmax (-27.7%) and diastolic function (LV–dp/dtmin (-27.6%), and elevated LVEDP (+82.2%; P<0.05, P<0.01), respectively. Dex significantly improved cardiac function (P<0.05, P<0.01), respectively. An increased heart rate (+19.6%, P<0.05) was seen in the CHF group, and it was also suppressed markedly by Dex treatment (Table 1).
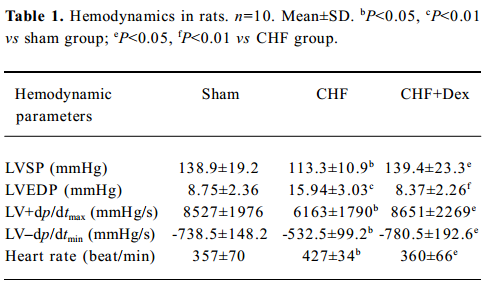
Full table
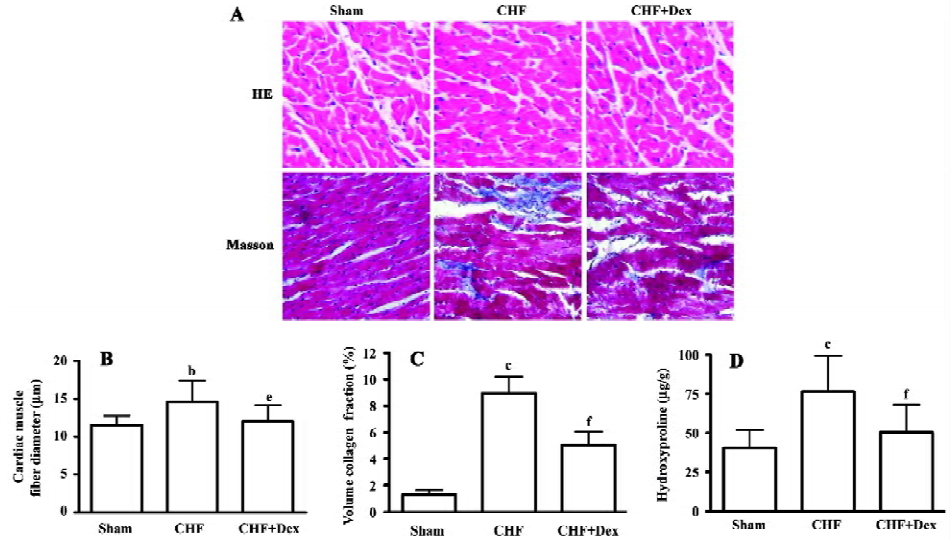
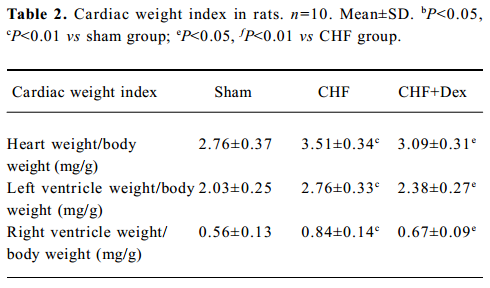
Remodeling of the myocardium was assessed morphologically and biochemically in CHF rats (Figure 1A; Table 2). The cardiac weight indice, a fraction of heart weight to body weight (HW/BW) and left and right ventricle weight to body weight (LVW/BW, RVW/BW, mg/g), increased by 27.2% (P<0.01), 36.0% (P<0.01), and 33.3% (P<0.01), respectively, compared with the sham group. Dex could effectively regress the increment in the cardiac weight indice significantly (P<0.05), compared with the CHF group (Table 2).
Full table
HE staining showed that the diameter of cardiac muscle fiber in the noninfarcted zone (free left ventricular wall) was significantly elongated in the CHF group compared with the sham group (P<0.05; Figure 1B). Masson staining showed an increase in blue staining indicating fibrosis of the myocardium in the rats with CHF.
The density of blue staining was markedly reduced after Dex treatment (Figure 1A). Semiquantitative scanning demonstrated that the fraction of collagen volume significantly increased by approximately 6 times compared with the sham group (P<0.01; Figure 1C). The level of myocardial hydroxyproline increased by 47.0% (P<0.01) in the CHF group compared with the sham group (Figure 1D). These abnormalities reflected significant cardiac remodeling during chronic heart failure and Dex treatment significantly reversed these changes induced by CHF (P<0.01).
Activity of cardiac enzymes in serum The marked elevations of GOT (+32.5%, P<0.01), GPT (+176.6%, P<0.01), LDH (+34.6%, P<0.05), and CPK (+22.2%, P<0.01) were observed in CHF group compared with the sham group. Dex treatment significantly reduced the elevation of the activities of the marker enzymes in serum (Table 3).
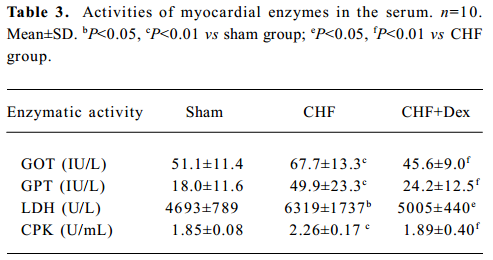
Full table
Oxidative stress and TNF-α mRNA expression There was significant changes in the activities of the redox system, SOD (-30.5%), MDA (+64.0%), GSH-Px (-19.6%), CAT (-34.7%), and XOD (+26.3%) in the serum in the CHF group compared to the sham group (P<0.01). Dex markedly reversed these changes in SOD, MDA, GSH-Px, CAT, and XOD induced by CHF (P<0.01, Table 4).
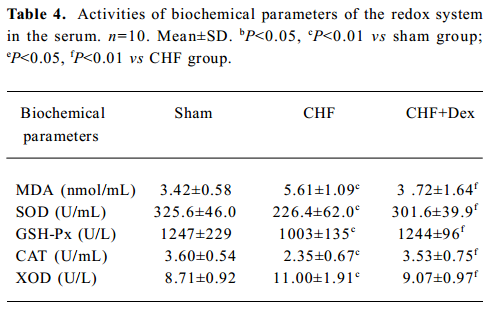
Full table
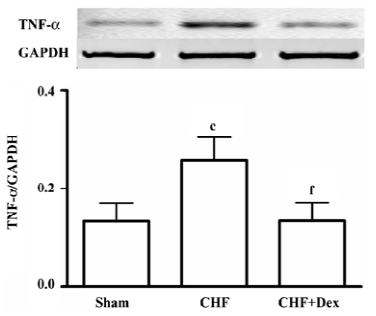
Compared with the sham group, TNF-α mRNA expression was up-regulated by 94.3% (P<0.01) in the CHF group. Dex treatment significantly suppressed the increase in TNF-α mRNA expression induced by CHF (P<0.01, Figure 2).
The mRNA expression and activities of MMP-2, MMP-9, TIMP-1, and TIMP-2 The mRNA expression of MMP-2 and MMP-9 was up-regulated by 112% (P<0.01) and 44.6% (P<0.01), respectively in the CHF group compared with the sham group. Dex significantly decreased upregulated mRNA expression of MMP-2 and MMP-9 induced by CHF (P<0.05 and P<0.01), respectively (Figure 3A, 3B). The mRNA expression of TIMP-1 and TIMP-2 was significantly down-regulated by 44.3% (P<0.05) and 44.1% (P<0.05), respectively in CHF group compared with the sham group. Dex markedly increased the down-regulation of the mRNA expression of TIMP-1 and TIMP-2 induced by CHF (P<0.01 and P<0.05, Figure 3C, 3D).
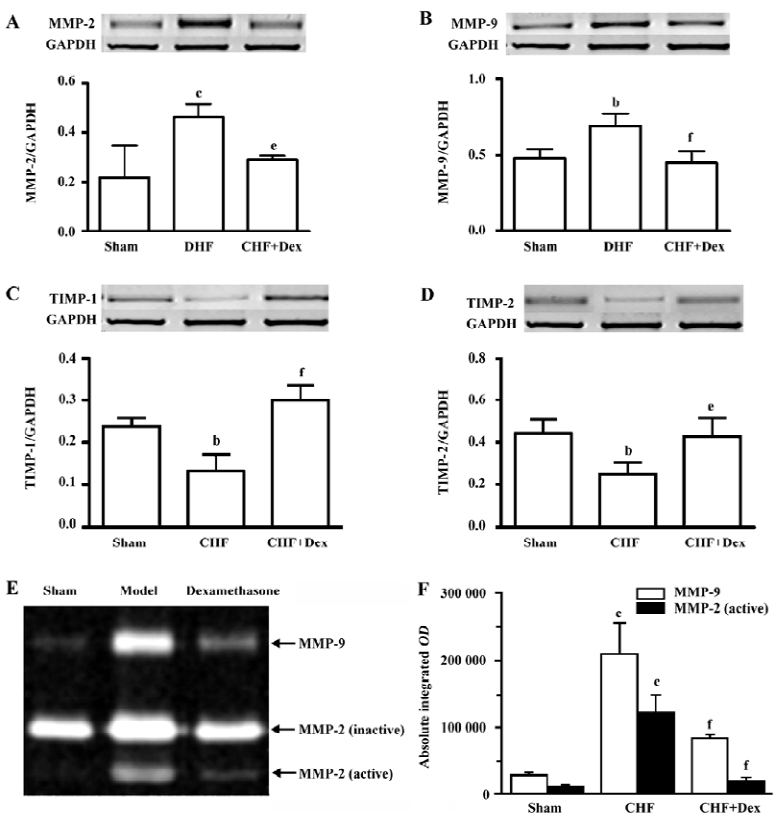
The lytic activities of MMP-2 and MMP-9 were significantly increased compared with the sham group (Figure 3E). Dex dramatically decreased elevated metalloproteinase activities induced by CHF (Figure 3F).
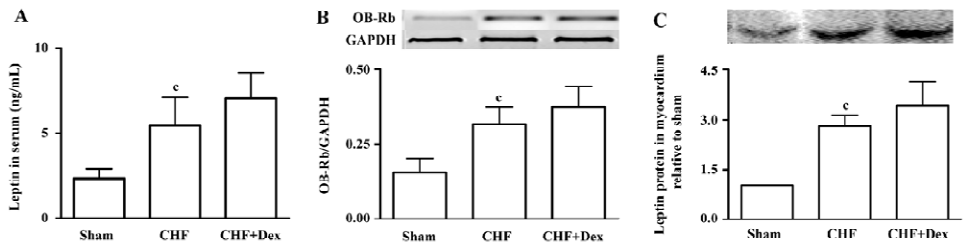
Serum leptin level and leptin receptor expression Hyperleptinemia was observed in the CHF group (P<0.01). The serum leptin level of the CHF group was 2 times higher compared with the sham group (Figure 4A). OB-Rb mRNA expression in the myocardium significantly increased by 104% in the CHF group compared with the sham group (P<0.01, Figure 4B). The leptin protein expression in the myocardium of the CHF group was also markedly up-regulated compared with the sham group (P<0.01; Figure 4C). There were no significant differences in the serum leptin level, mRNA expression of OB-Rb in myocardium, and leptin protein expression between the Dex-treated CHF group and the CHF group (P>0.05).
Discussion
Leptin, a 16 kDa peptide, was produced primarily by adipocytes. It is referred to as an anti-obesity hormone due to its inhibitory effects on food intake and stimulating effects on energy expenditure. The heart is a site of leptin production and its action is mediated by a group of a soluble form of leptin receptors[1]. One of the isoforms of the receptor, known as the long form (OB-Rb), is considered to represent the full signaling isoform of the leptin receptor (OB-Ra, OB-Rb, OB-Rc, OB-Rd, OB-Re, and OB-Rf). In this study, we observed that an upregulation of serum leptin levels, leptin protein expression, and mRNA expression of leptin receptor OB-Rb was associated with impaired cardiac function after CHF, which was in agreement with hyperleptinemia following myocardial infarction[5], and the upregulated leptin receptor in the myocardium of failing hearts[6].
The upregulation of TNF-α found in CHF group was linked with an increase in serum leptin level. Both TNF-α and leptin inhibited cardiac contractile function independently and synergistically[17]. The production of TNF-α in injured myocardium was promoted by leptin, so the 2 cytokines co-contributed to the morbidity of CHF[18].
Change in the extracellular matrix in the myocardium, primarily responsible for cardiac remodeling, is mediated by an increase in activities of MMP, which can collectively degrade all structural extracellular matrix proteins of the failing hearts[20]. During the process of CHF, MMP are initially activated to reduce wall stress and allow dilation against an increased workload induced by increasing fibril collagen degradation. In this study, the expression and activities of MMP were upregulated significantly in the CHF group in association with a reduction in mRNA expression of the endogenous inhibitor TIMP-1 and TIMP-2 in the myocardium. Cardiac structure changes are attributed to abnormal expression of MMP-2, MMP-9, TIMP-1 and TIMP-2. Interstitial fibrosis is commonly exhibited at the end stage of dilated, ischemic, and valvular cardiomyopathy, but it can be discriminated by variations in volume density of type III collagen and laminin, and MMP in the myocardium[21]. Cardiac function of the chronic infarcted heart could be deteriorated by an activated MMP-2, MMP-9, and reduced TIMP-1 and TIMP-2. In the myocardium, the ultrastructural collagen, initially degraded by MMP, is replaced by poorly-structured collagen; then cardiac remodeling takes place. Some MMPs leak into the blood circulation after myocardial infarction, so the serum MMP level can be used to predict the occurrence of congestive heart failure in patients with myocardial infarction during a 2-year follow up period. It is interesting to find that elevated level of MMP-9, rather than the levels of TNF-α, the C-reactive protein, or MMP-2, may be a significant risk factor for the late onset of CHF in patients with acute myocardial infarction[22]. The upregulation of MMP-2 in the myocardium is significant in CHF patients and contributes to cardiac remodeling; however, on the other hand the up-regulated MMP-2 had beneficial effects on CHF patients. Cardiomyopathy induced by over-expression of TNF-α in transgenic mice was associated with cardiac remodeling and heart failure and up-regulation of MMP-2. In MMP-2 knockout (MMP-2 [–/–]) mice, the zymographic activity of MMP-2 is completely abolished; however, cardiac function is worsened and the survival time is shortened compared with the wild-type MMP-2 mice. Thus, the upre-gulation of MMP-2 in the process of CHF could be a protective factor in TNF-α-induced cardiomyopathy[23]. We observed that Dex attenuated myocardial remodeling and inhibited the MMP activities which coincided with previous findings[24].
Oxidative stress is found predominantly in CHF and contributes to myocardial remodeling by activating MMP-2 and MMP-9 and depressing TIMP-1 and TIMP-2 expression[25]. TNF-α plays an important role in oxidative stress in the pathogenesis of myocardial remodeling and chronic cardiac failure. TNF-α induces reactive oxygen species (ROS) production and is involved in structural changes in hypertrophy[26]. The stimulation of ROS was reflected by the increase in the level of malondialdehyde (MDA) and the activity of oxidant enzyme (XOD) and the decrease in the level of anti-oxidant enzymes (SOD, GSH-px, and CAT) in the serum. Oxidative stress is associated with myocardial contractile dysfunction and structural remodeling. ROS are also involved in the process of apoptosis of cardio-myocytes and interstitial fibrosis, both of which contributed to the development of myocardial structural damage. Our present study showed that Dex decreased ROS activities in the serum as well as TNF-α mRNA expression in the myocardium.
We demonstrated that Dex ameliorated CHF induced by coronary artery ligation in rats. The findings are in agreement with several reports that Dex protects the heart from ischemia/reperfusion injury[27,28].
The leptin receptor OB-Rb belongs to the gp130 family of cytokine receptors and acts primarily through the activation of the Janus kinase (JAK) family of cytoplasmic tyrosine kinases, which in turn activate transcription factors of the signal transducer, and is member of an activator of the transcription (STAT) family[29]. Leptin stimulates cardiomyocyte hypertrophy through various intracellular signaling cascades including the JAK/STAT, p38 mitogen activated protein kinase (MAPK), extracellular signal regulated kinase (ERK), and the PI3K/Akt pathway[10,30]. Glucosteroids rapidly reduce leptin-induced JAK/STAT signaling and a specific MEK inhibitor PD98059 blocks the inhibitory effects of glucosteroids on leptin-induced JAK/STAT activation in Huh7 cells[13], without changing leptin binding to the cells. Thus, these support our findings that the glucosteroid Dex could rapidly block leptin signaling pathway, possibly by significant inhibition of the leptin-induced JAK/STAT pathway with no effect on the serum levels, and the upregula-tion of leptin and its receptors in the myocardium. The cytopro-tective effect by Dex is related to de novo protein synthesis[31].
Dexamethasone injection and feeding increased plasma leptin concentrations in dogs. In addition, dexamethasone administration enhanced the effect of feeding on increases in plasma leptin concentrations. Daily oral administration of prednisolone (1 or 2 mg/kg) did not affect plasma leptin concentrations in dogs[32]. Dex significantly increased OB-Rb mRNA expression, but leptin inhibited OB-Rb mRNA expression[33]. This suggests that glucocorticoids as well as leptin itself had regulatory effects on gene expression of leptin receptors. Dex stimulates the upregulation of leptin in combination with either TNF-α[34] or insulin[35], so, these provide an explanation for improvement of CHF by Dex. Dex upregulates leptin and its receptors, but suppressed it signaling pathway. Thus it is likely when other inflammatory factors contributing to the development of CHF are suppressed by Dex, the leptin could remain unchanged.
Leptin increases sympathetic activation[33], generates ROS, and upregulates TNF-α, MMP-2, and MMP-9 in the myocardium. However, it is a phenomenon of leptin resistance which is associated with hyperleptinemia with no biological effects[33]. It remains unclear in biology of leptin and it is still uncertain whether leptin is a friend or a foe to the cardiovascular system[18].
In conclusion, hyperleptinemia and the upregulation of the leptin protein and receptors do not substantially contribute to CHF. Leptin seems to be only a marker in CHF, which can be greatly relieved by Dex; however, hyperleptinemia and the upregulation of the leptin remain unchanged.
References
- Purdham DM, Zou MX, Rajapurohitam V, Karmazyn M. Rat heart is a site of leptin production and action. Am J Physiol Heart Circ Physiol 2004;287:H2877-84.
- Wannamethee SG, Tchernova J, Whincup P, Lowe GD, Kelly A, Rumle A, et al. Plasma leptin: associations with metabolic, inflammatory and haemostatic risk factors for cardiovascular disease. Atherosclerosis 2006. [Epub ahead of print].
- Piatti PC, Di Mario LD, Monti G, Fragasso F, Sgura A, Caumo A, et al. Association of insulin resistance, hyperleptinemia, and impaired nitric oxide release with in-stent restenosis in patients undergoing coronary stenting. Circulation 2003;108:2074-81.
- Thakur V, Richards R, Reisin E. Obesity, hypertension, and the heart. Am J Med Sci 2001;321:242-8.
- Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, et al. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail 2003;5:33-40.
- Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure-overloaded human myocardium during heart failure progression. J Am Coll Cardiol 2004;44:1609-18.
- Seddon M, Looi AYH, Shah M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2006.2. [Epub ahead of print].
- Castellucci MR, De Matteis A, Meisser R, Cancello V, Monsurro D. Leptin modulates extracellular matrix molecules and metalloproteinases: possible implications for trophoblast invasion. Mol Hum Reprod 2000;6:951-8.
- Li L, Mamputu JC, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes 2005;54:2227-34.
- Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004;110:1269-75.
- Conraads VM, Denollet J, De Clerck LS, Stevens WJ, Bridts CC, Vrints J. Type D personality is associated with increased levels of tumour necrosis factor (TNF)-alpha and TNF-alpha receptors in chronic heart failure. Int J Cardiol 2006;97:970-3.
- Finck BN, Johnson RW. Anti-inflammatory agents inhibit the induction of leptin by tumor necrosis factor-alpha. Am J Physiol Regul Integr Comp Physiol 2002;282:R1429-35.
- Ishida-Takahashi R, Uotani S, Abe T, Degawa-Yamauchi M, Fukushima T, Fujita N, . Rapid inhibition of leptin signaling by glucocorticoids in vitro and in vivo. J Biol Chem 2004; 279: 19 658–64.
- Machuca C, Mendoza-Milla C, Cordova E, Mejia S, Covarrubias L, Ventura J, et al. Dexamethasone protection from TNF-alpha-induced cell death in MCF-7 cells requires NF-kappaB and is independent from AKT. BMC Cell Biol 2006;7:9.
- Bruder ED, Jacobson L, Raff H. Plasma leptin and ghrelin in the neonatal rat: interaction of dexamethasone and hypoxia. J Endocrinol 2005;185:477-84.
- Fried SK, Ricci MR, Russell CD, Laferrere B. Regulation of leptin production in humans. J Nutr 2000;130:S3127-31.
- Ren JD, Relling P. Interaction between tumor necrosis factor-alpha and leptin-induced inhibition of cardiac contractile function in isolated ventricular myocytes. Cytokine 2005;32:213-8.
- Ren J. Leptin and hyperleptinemia – from friend to foe for cardiovascular function. J Endocrinol 2004;181:1-10.
- Nagaya N, Uematsu M, Kojima M, Ikeda Y, Yoshihara F, Shimizu W, et al. Chronic administration of ghrelin improves left ventricular dysfunction and attenuates development of cardiac cachexia in rats with heart failure. Circulation 2001;104:1430-5.
- Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, et al. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation 2006;113:2089-96.
- Herpel E, Singer S, Flechtenmacher C, Pritsch M, Sack FU, Hagl S, et al. Extracellular matrix proteins and matrix metallopro-teinases differ between various right and left ventricular sites in end-stage cardiomyopathies. Virchows Arch 2005;446:369-78.
- Wagner DR, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail 2006;12:66-72.
- Matsusaka H, Ikeuchi M, Matsushima S, Ide T, Kubota T, Feldman AM, et al. Selective disruption of MMP-2 gene exacerbates myocardial inflammation and dysfunction in mice with cytokine-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 2005;289:H1858-64.
- Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circulation 2002;105:1983-8.
- Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res 2006;98:271-9.
- Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol 2004;287:H1813-20.
- Valen G, Kawakami T, Tahepold P, Dumitrescu A, Lowbeer C, Vaage J. Glucocorticoid pretreatment protects cardiac function and induces cardiac heart shock protein 72. Am J Physiol Heart Circ Physiol 2000;279:H836-43.
- Varga E, Nagy N, Lazar J, Czifra G, Bak I, Biro T, et al. Inhibition of ischemia/reperfusion-induced damage by dexamethasone in isolated working rat hearts: the role of cytochrome c release. Life Sci 2004;75:2411-23.
- Ahima RS, Flier JS. Leptin. Annu Rev Physiol 2000;62:413-37.
- Rajapurohitam V, Gan XT, Kirshenbaum M, Karmazyn LA. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res 2003;93:277-9.
- Tosaki A, Koltai M, Joo F, Adam G, Szerdahelyi P, Lepran I, et al. Actinomycin D suppresses the protective effect of dexamethasone in rats affected by global cerebral ischemia. Stroke 1985;16:501-5.
- Nishii N, Takasu M, Ohba Y, Maeda S, Kitoh K, Ohtsuka Y, et al. Effects of administration of glucocorticoids and feeding status on plasma leptin concentrations in dogs. Am J Vet Res 2006;67:266-70.
- Luo JD, Zhang GS, Chen MS. Leptin and cardiovascular diseases. Drug News Perspect 2005;18:427-31.
- Trujillo ME, Lee MJ, Sullivan S, Feng J, Schneider SH, Greenberg AS, et al. Tumor necrosis factor alpha and glucocorticoid synergistically increase leptin production in human adipose tissue: role for p38 mitogen-activated protein. Clin Endocrinol Metab 2006;91:1484-90.
- Ramsay TG, Richards MP. Hormonal regulation of leptin and leptin receptor expression in porcine subcutaneous adipose tissue. J Anim Sci 2004;82:3486-92.