
Activation of PI3-K/Akt pathway for thermal preconditioning to protect cultured cerebellar granule neurons against low potassium-induced apoptosis1
Introduction
Neuronal apoptosis sculpts the developing brain. How-ever, inappropriate apoptosis has been suggested to play a potentially important role in the pathogenesis of various neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease[1,2]; it has also been implicated in other types of neurological disorders such as cerebral ischemia[3]. It is of a great importance to establish therapeutic strategies to combat apoptosis-induced damage to the central nervous system and to elucidate the underlying mechanisms.
Thermal preconditioning (TP) can result in the acquisition of tolerance. It means tissues and cells are subjected to sub-lethal heat stress to obtain the ability to withstand subsequent usually lethal stresses. Previous studies have shown that thermal preconditioning is able to promote neuronal survival under various types of stress. Although the underlying mechanisms are usually ascribed to the increased expression of one or more heat shock proteins (HSP)[4], some studies have documented that the overexpression of HSP fails to rescue the neurons from apoptosis induced by heat or ischemic stress[5]. So the precise mechanisms are still unclear.
Some evidence suggests that the activation of Akt is enhanced immediately after heat stress[6,7]. Akt, a serine/threonine kinase, plays a critical role in regulating neuronal cell survival responses. Akt is a downstream effector of phosphatidylinositol 3-kinase (PI3-K), and the phosphorylated form has been demonstrated to maintain cell survival by inactivating several apoptosis effectors such as BAD, forkhead transcription factors, caspase 9 and glycogen synthase kinase 3β (GSK3β)[8,9]. Moreover, the overexpression of constitutive Akt by the transfection of active Akt cDNA into cells can rescue the cells from apoptosis induced by various types of stress[10]. These findings have led to the hypothesis that a rise in the activation level of Akt contributes to the TP-induced neuroprotective effect. However, how the activation of the PI3-K/Akt pathway is involved in the TP-induced neuroprotective effect has not been examined.
Cultured cerebellar granule neurons (CGN) from early postnatal rats represent a highly homogeneous neuron population. CGN survive and develop characteristics of mature CGN when maintained in depolarizing concentrations of potassium (25 mmol/L) medium, but undergo apoptosis characterized by chromatin condensation and DNA fragmentation when cultured in physiological low potassium (5 mmol/L) conditions[11]. The low potassium-induced cerebellar granule neuronal apoptotic paradigm has been proposed as a suitable in vitro model for studying the mechanisms of neuronal apoptosis involved in many neurodegenerative diseases due to its excellent stability.
In this study, we investigated whether the activation of the PI3-K/Akt pathway was required for thermal preconditioning to protect CGN against apoptosis induced by low potassium. Furthermore, we explored the possibility of a link between the upregulated HSP70 expression and Akt activation in the acquisition of neuroprotection induced by thermal preconditioning. We provide a new pathway to elucidate the mechanisms of the acquisition of tolerance induced by thermal preconditioning.
Materials and methods
Materials All chemicals, unless otherwise noted, were purchased from Sigma Chemicals (Sigma-Aldrich, St Louis, MO, USA). LY294002 was purchased from Calbiochem (San Diego, CA, USA). Primary antibodies against total Akt, phospho-Ser473Akt, GSK3β, or phospho-Ser9GSK3β, secondary antibodies, including horseradish peroxidase (HRP)-linked anti-rabbit antibody and HRP-linked anti-mouse antibody and HRP-conjugated anti-biotin antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). The primary antibody against HSP70 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The primary antibody against β-actin was from Neomarkers (Fremont, CA, USA).
Cell culture and treatment Rat cerebellar granule neurons were prepared as described by Yan et al[12]. The cerebella were obtained from Sprague-Dawley rat pups (postnatal 7–8 d). They were mechanically disrupted, then trypsinized with 0.025% trypsin (including 0.01% DNase I) for 15 min at 37 °C. Trypsin digestion was stopped by the addition of trypsin inhibitor (0.05%). After trituration, cerebellar granule neurons were plated in poly-lysine-coated 24-well or 35-mm culture plates at a density of (1.5–1.8)×109 cells/L in basal modified Eagle’s (BME) medium containing 10% fetal bovine serum, 25 mmol/L KCl, 2 mmol/L glutamine, 100 kU/L penicillin, and 100 mg/L streptomycin, and incubated at 37 °C with 5% CO2 in a humidified chamber. Cytosine arabinoside (10 µmol/L) was added to the medium 24 h after plating to arrest the growth of non-neuronal cells. D-glucose (5 mmol/L) was added to the cultures on d 6. On d 8, in vitro CGN were rinsed twice and maintained in 25K medium (containing 25 mmol/L serum-free KCl) to be used in the experiments. To induce neuronal apoptosis, CGN were switched to 5K medium (containing 5 mmol/L serum-free KCl) for 24 h.
TP CGN were incubated at 43.5 °C with 5% CO2 in a humidified chamber for 90 min and recovered at 37 ℃ with 5% CO2 in a humidified chamber for 1 h before low potassium (5K) treatment[13].
Drug treatment The PI3-K inhibitor, LY294002, was dissolved in dimethyl sulfoxide (DMSO) as a 1000×50 µmol/L stock solution and then added to 25K medium; the final concentration of LY294002 was 5,10, or 20 µmol/L according to different experiments. The 25K medium with 0.2% DMSO was used as the control. In the preliminary experiments, we found that DMSO concentrations up to 0.3% did not affect neuronal viability of CGN within 120 h of exposure. However, unless otherwise stated, the final concentration of DMSO was 0.2%. LY294002 was added to the medium 1 h before thermal preconditioning.
To examine the role of PI3-K/Akt in the thermal preconditioning-mediated survival protection of CGN, we designed an experiment on CGN with a specific PI3-K inhibitor, LY294002. Thus, CGN were divided into 5 groups as follows: control (25K group), 5K treatment (5K group), thermal preconditioning+5K treatment (H+5K group), 20 µmol/L LY294002+thermal preconditioning+5K treatment (LY+H+5K group), and the 20 μmol/L LY294002+25K (LY+25K group) groups. The LY+H+5K group was treated with LY294002 for 1 h before thermal preconditioning (at 43.5 °C for 90 min, then at 37 °C for 1 h) and was then switched to 5K medium. The LY+25K group was treated with LY294002 for 3.5 h and was then switched to 25K medium. Cells cultured in 24-well plates were used to perform MTT assay or fluorescein diacetate (FDA) staining for evaluating survival cells. Four wells were used for each group. Cells cultured in 35-mm plates were used for Hoechst 33258 staining, agarose gel electro-phoresis, or Western blot analysis. All experimental procedures were performed according to the Biomedical Security Rule.
MTT assay and FDA staining for evaluating survival cells 3-(4,5-Dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) assay was performed as described previously[14]. Briefly, the neurons in the 24-well plates were cultured and treated according to the above methods. MTT was added to the medium at a final concentration of 1 mg/mL and the cells were incubated at 37 °C for 4 h. Then the medium was removed. DMSO was then applied to the wells to dissolve the formazan crystal. The absorbance of the samples was measured at a wavelength of 570 nm with 630 nm as reference wavelength using the Universal Microplate Reader (ELX 800, Bio-Tek, Winooski, Vermont, USA). Values are expressed as a percentage of control cultures for each experiment. For FDA staining, as described previously[12], CGN were incubated with fluorescein diacetate (10 mg/L) at 37 ℃ for 5 min and then examined and randomly photographed by fluorescence microscope at ×200 magnification. The number of neurons was counted from the photos by a blind observer. Neuronal survival was calculated by the following formula: neuronal survival (%)=(survival cells in the treated group/survival cells in the control group)×100%.
Hoechst 33258 staining for detecting chromatin condensation Chromatin condensation was detected by nucleus staining with Hoechst 33258 as described previously[12]. CGN grown in the 35-mm dishes were washed with ice-cold phosphate-buffered saline (PBS) and fixed with 4% formaldehyde in PBS. Cells were then stained with Hoechst 33258 (5 mg/L) for 10 min at room temperature. Nuclei were visualized using a fluorescence microscope at ×1000 magnification. In this way, apoptotic CGN would be stained into bright blue because of their chromatin condensation, while normal CGN were stained to a slight blue colour.
Agarose gel electrophoresis for detecting DNA fragmentation Genomic DNA was isolated from neurons grown in poly-lysine-coated 35-mm tissue culture dishes. Cells were rinsed with BME, scraped, collected in ice-cold PBS, and centrifuged at 5000×g for 5 min. The pellet was lysed in 600 μL of a buffer consisting of 10 mmol/L Tris-HCl, 10 mmol/L edetic acid and 0.2% Triton X-100 (pH 7.5). After 15 min on ice, the lysate was centrifuged at 12 000×g at 4 ºC for 10 min. The supernatant was extracted first with phenol and then with phenol-chloroform: isoamylalcohol (24:1). The aqueous phases were mixed with a 1/10 volume of 3 mol/L sodium acetate (pH 5.2) and an equal volume of ice-cold isopropanol for 24 h at -20 ℃. The pellet was washed with 70% ethanol, air-dried and dissolved in 15 μL of Tris-HCl 10 mmol/L plus 1 mmol/L edetic acid (pH 7.5). After digesting RNA with RNase A (0.6 g/L, at 37 ℃ for 30 min), the sample was electrophoresed in 1.5% agarose gel with TBE buffer. DNA was visualized by ethidium bromide staining.
Western blot analysis Western blot analysis was performed as described previously and slightly modified[15]. In brief, neurons were harvested in a cell lysis buffer (Pierce, Rockford, IL, USA). After incubation on ice for 15 min and centrifugation at 18 000×g at 4 ℃ for 10 min, whole protein concentrations were determined by BCA assay (Pierce, USA), using bovine serum albumin as a standard. Cell lysates were diluted in the SDS sample buffer, and the mixture was boiled for 5 min. The protein (30 µg) was separated on a 12% SDS-PAGE. Proteins were transferred to the nitrocellulose mem-branes. The membranes were incubated for 60 min at room temperature in Tris-buffered saline containing 0.05% Tween-20 (TBST) and 5.% nonfat dry milk (NFDM) and then overnight at 4 ºC with specific antibodies for phospho-Ser473Akt (1:1000), phospho-Ser9 GSK3β (1:1000), Akt (1:1000), GSK3β (1:1000), or HSP70 (1:500). The membranes were then washed in TBST (3×5 min) and incubated for 60 min in TBST/1%NFDM containing a 1:2000 dilution of a HRP-conjugated secondary antibody. This was followed by washes (3×15 min) in TBST and subsequently was developed with an enhanced chemiluminescence system (Pierce, USA). If necessary, the blots were then incubated in stripping buffer (67.5 mmol/L Tris, pH 6.8, 2% SDS, and 0.7% β mercapto-ethanol) at 50 ºC for 30 min and then reprobed with antibodies against β-actin (1:1000) as loading controls. Quantification was performed with the Bio-Rad Quantity One software (Hercules, CA, USA). All data from 3 independent experiments were expressed as the ratio to optical density values of the corresponding controls for the statistical analyses.
Statistical analysis Data are presented as mean±SD from independent experiments. For the Western blots, each experiment was repeated at least 3 times, and in all cases, the same results were obtained. Statistical analysis of data was performed by Student’s t-test. P<0.05 was considered significant.
Results
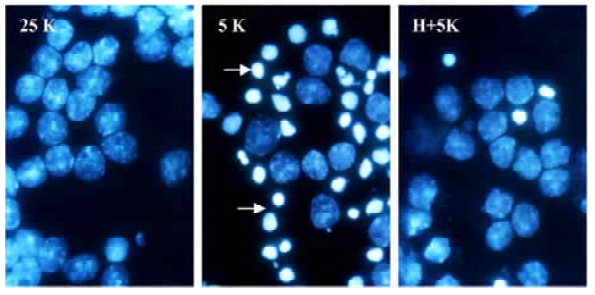
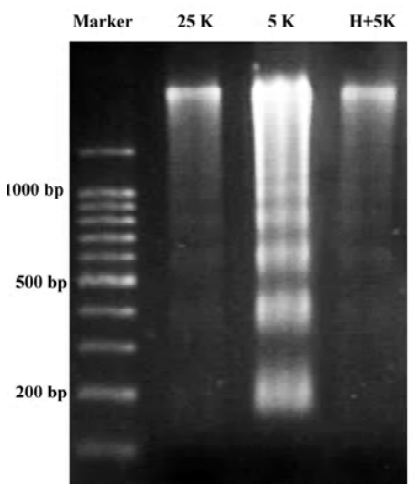
TP protects CGN against apoptosis induced by low potassium To investigate the effects of TP on apoptosis of CGN induced by low potassium, at 24 h after 5K treatment, CGN were stained by Hoechst 33258, a classical way of identifying apoptotic cells, to test nuclei morphology of neurons. The results indicated that nuclei of most CGN in the 5K group were stained bright blue; the nuclei of most CGN in 25K group and H+5K group showed a slight blue (Figure 1). Another biochemical feature of apoptosis, DNA fragmentation, was also tested. We found that CGN exposed to 5K medium for 24 h showed a typical internucleosomal DNA fragmenta-tion, while no ladders of oligonucleosomal length DNA were detected in CGN of the 25K group and the H+5K group (Figure 2). The results demonstrated that TP protected CGN against apoptosis induced by low potassium.
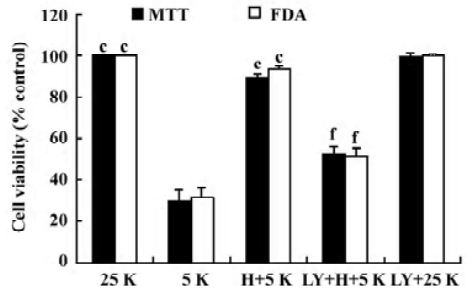
LY294002 inhibits the neuroprotective effect on CGN induced by TP To examine the role of the PI3-K/Akt pathway in the TP-mediated protection of CGN, we performed an experiment on CGN with LY294002. Thus, CGN were divided into 5 groups as described in Materials and methods. At 24 h after the treatment with 5K, cell viability was determined by MTT assay and FDA staining. Compared with the H+5K group, cell viability in the LY294002+H+5K group decreased significantly (P<0.01); however, compared with the 25K group, cell viability in the LY294002+25K group did not decrease (P>0.05; Figure 3). The results revealed that LY294002 potently inhibited the neuroprotective effect of TP against low potassium-induced apoptosis of CGN.
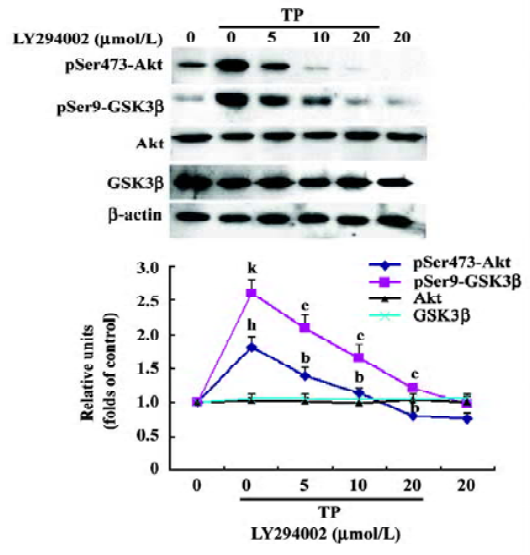
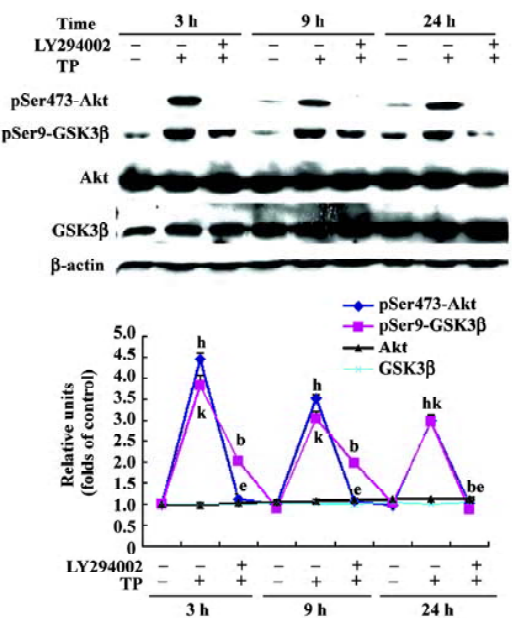
TP induces Akt and GSK3β phosphorylation in a PI3-K-dependent manner To determine whether the activation of the PI3-K/Akt pathway was involved in the neuroprotective activity of TP against low potassium-induced apoptosis of CGN, after TP following the pretreatment of different concentrations of LY294002 (5, 10, and 20 µmol/L) for 1 h, cells were harvested for immuno-blotting. TP resulted in a robust activation of Akt and an inactivation of GSK3β as indicated by increased phospho-Ser473-Akt and increased phospho-Ser9-GSK3β (P<0.05), whereas total Akt and total GSK3β did not change (P>0.05). Furthermore, LY294002 decreased TP-induced phosphorylation of Akt and GSK3β dose-dependently (P<0.05, Figure 4). The results revealed that TP activated Akt and subsequently inactivated GSK3β via PI3-K.
TP induced Akt and GSK3β phosphorylation in 5K cultures If the activation of PI3-K/Akt pathway by TP protects CGN against apoptosis induced by low potassium at 24 h, its activation may persist for 24 h. So cells of the 5K group, the H+5K group, and the LY294002+H+5K group were harvested 24 h after the treatment of 5K for immunoblotting. Compared with the 5K group, TP induced Akt and GSK3β phosphorylation persistently. Even at the time point of 24 h after exposure to 5K, the levels of phospho-Ser473 Akt and phospho-Ser9 GSK3β of CGN in the H+5K group were still much higher than that of CGN in the 5K group (P<0.05). Moreover,LY294002 significantly inhibited Akt and GSK3β phosphorylation induced by TP in 5K cultures (P<0.05; Figure 5).
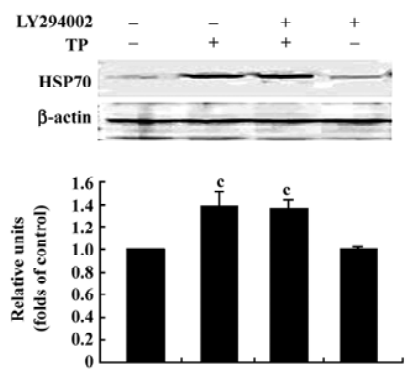
Inhibition of the PI3-K/Akt pathway did not affect the upregulated HSP70 expression induced by TP The above results show that the activation of the PI3-K/Akt pathway was required for TP to protect CGN against apoptosis induced by low-potassium. Our previous report showed that IP-induced HSP70 expression was also involved in TP-mediated anti-apoptotic action on CGN[13]. To explore the possibility of a link between the upregulated HSP70 expression and the activation of Akt in the acquisition of neuroprotection induced by TP, we examined the effect of LY294002 (20 µmol/L) on the upregulated HSP70 expression induced by TP. The result revealed that TP apparently increased the levels of HSP70 (P<0.01); however, LY294002 (20 µmol/L) had no effect on the increased levels of HSP70 induced by TP (P>0.05; Figure 6).
Discussion
Neuronal apoptosis resulting from a variety of severe stresses has been emphasized in the pathogenesis of numerous neurodegenerative diseases. Mobilizing intrinsic defensive mechanisms to deal with apoptosis is important for protecting neurons. TP leads to the phenomenon of thermal tolerance in which sub-lethal heat stress induces robust protection against subsequent, potentially lethal heat stress[16]. Much evidence has shown that TP not only produces thermal tolerance[17], but also provides the brain and neurons with a protective response against subsequent various lethal insults such as ischemia and hypoxia injury[18,19], glutamate excitotoxicity[20,21] and toxicity of 1-methyl-4-phenylpyridinium ion[22] etc. The present study also shows that TP protects CGN against low potassium-induced apoptosis.
Usually the production of HSP following TP, such as the HSP70 protein which misfolded or damaged chaperone, is regarded as one of the critical mechanisms used to bolster cellular defenses to support neuronal survival in the face of potentially lethal conditions[23,24]. However, the other survival-promoting mechanisms besides HSP may also be involved in the TP-mediated neuroprotective effect. The PI3-K/Akt signaling pathway is considered a classic pro-survival pathway. PI3-K phosphorylates Akt to give rise to phosphorylated Akt. The activation of Akt supports cell survival and counteracts apoptotic signaling, a part of which relies on the inactivation of GSK3β by converting phosphorylated GSK3β[25]. In the present study, TP induced the activation of Akt and the inactivation of GSK3β in a PI3-K-dependent manner. The inhibition of the PI3-K/Akt pathway by LY294002 reduced the neuroprotective effect of TP against low potassium-induced neuronal death; moreover, the activation of Akt and the inactivation of GSK3β by TP persisted for 24 h during the treatment of low potassium. So our findings support the hypothesis that the activation of the PI3-K/Akt pathway by TP protects CGN against apoptosis induced by low potassium. Although previously there were reports of heat stress modulating the activity of the PI3-K/Akt pathway[7], the present study demonstrates that the activation of the PI3-K/Akt pathway is required for the TP-induced neuro-protective effect. Our findings support that TP is a potential neuroprotective means for neurodege-nerative diseases since the activation of the PI3-K/Akt pathway is widely involved in anti-apoptotic action in these diseases.
It has been reported that the upregulated HSP70 expression induced by TP participates in the TP-mediated neuro-protective effect[13]. Is there a link between the upregulated HSP70 expression and the activation of PI3-K/Akt pathway in the acquisition of neuroprotection induced by TP? Considering that neuroprotection by Akt can be mediated through the downstream induction of HSP70 expression[26], and in renal cell carcinoma (RCC4) cells LY294002 largely attenuates increased HSP70 expression toward heat treatment[27], we speculate in this assay that HSP70 might be the downstream target of Akt and that LY294002 attenuates TP-induced neuroprotection, probably through inhibiting increased HSP70 expression. Our data reveals that TP resulted in the upregulated HSP70 expression, but LY294002 exerted no effects on the TP-induced upregulated HSP70 expression. This means that neuroprotection by the IP-induced PI3-K/Akt pathway activation was not mediated through the downstream induction of HSP70 expression in the present study. Contradictory evidence concerning a link between Akt activity and HSP70 expression may be due to cell-specific differences in the signaling pathways that are recruited. So the possibility of a link between Akt activity and HSP70 expression in the acquisition of neuroprotection induced by thermal preconditioning remains to be clarified.
References
- Sastry PS, Subba RK. Apoptosis and the nervous system. J Neurochem 2000;74:1-20.
- Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci 2000;23:73-7.
- Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 min focal cerebral ischemia in rats. Stroke 1995;26:1252-8.
- Ohtsuka K, Suzuki T. Roles of molecular chaperones in the nervous system. Brain Res Bull 2000;53:141-6.
- Mailhos C, Howard MK, Latchman DS. Heat shock proteins hsp90 and hsp70 protect neuronal cells from thermal stress but not from programmed cell death. J Neurochem 1994;63:1787-95.
- Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-Kinase and glycogen synthase kinase-3 in the regulation of HSF-1 activity. J Neurochem 2000;75:2401-8.
- Maroni P, Bendinelli P, Tiberio L, Rovetta F, Piccoletti R. In vivo heat-shock response in the brain: signalling pathway and transcription factor activation. Brain Res Mol Brain Res 2003;119:90-9.
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 2001;26:657-64.
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 2002;12:275-8.
- Goswami R, Kilkus J, Dawson SA, Dawson G. Overexpression of Akt (protein kinase B) confers protection against apoptosis and prevents formation of ceramide in response to pro-apoptotic stimuli. J Neurosci Res 1999;57:884-93.
- D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 1993; 90: 10 989–93.
- Yan GM, Ni B, Weller M, Wood KA, Paul SM. Depolarization or glutamate receptor activation blocks apoptotic cell death of cultured cerebellar granule neurons. Brain Res 1994;656:43-51.
- Chen LJ, Su XW, Qiu PX, Huang YJ, Yan GM. Thermal preconditioning protected cerebellar granule neurons of rat by modulating HSP70 expression. Acta Pharmacol Sin 2004;25:458-61.
- Castoldi AF, Barni S, Randine G, Costa LG, Manzo L. Ethanol selectively interferes with the trophic action of NMDA and carbachol on cultured cerebellar granule neurons undergoing apoptosis. Brain Res Dev Brain Res 1998;111:279-89.
- Zhao LZ, Su XW, Huang YJ, Qiu PX, Yan GM. Activation of c-Jun and suppression of phospho-p44/42 were involved in diphenylhydantoin-induced apoptosis of cultured rat cerebellar granule neurons. Acta Pharmacol Sin 2003;24:539-48.
- McAlister L, Finkelstein DB. Heat shock proteins and thermal resistance in yeast. Biochem Biophys Res Commun 1980;93:819-24.
- Wang JL, Ke DS, Lin MT. Heat shock pretreatment may protect against heatstroke-induced circulatory shock and cerebral ischemia by reducing oxidative stress and energy depletion. Shock 2005;23:161-7.
- Ikeda T, Ikenoue T, Xia XY, Xia YX. Important role of 72-kd heat shock protein expression in the endothelial cell in acquisition of hypoxic-ischemic tolerance in the immature rat. Am J Obstet Gynecol 2000;182:380-6.
- Basaran M, Kafali E, Sayin O, Ugurlucan M, Us MH, Bayindir C, et al. Heat stress increases the effectiveness of early ischemic preconditioning in spinal cord protection. Eur J Cardiothorac Surg. 2005;28:467-72.
- Kwong JM, Lam TT, Caprioli J. Hyperthermic pre-conditioning protects retinal neurons from N-methyl-D-aspartate (NMDA)-induced apoptosis in rat. Brain Res 2003;970:119-30.
- Rordorf G, Koroshetz WJ, Bonventre JV. Heat shock protects cultured neurons from glutamate toxicity. Neuron 1991;7:1043-51.
- Fan GH, Qi C, Chen SD. Heat shock proteins reduce toxicity of 1-methyl-4-phenylpyridinium ion in SK-N-SH cells. J Neurosci Res 2005;82:551-62.
- Edwards MJ. Apoptosis, the heat shock response, hyperthermia, birth defects, disease and cancer. Where are the common links? Cell Stress Chaperones 1998;3:213-20.
- Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med 1999;31:261-71.
- Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 1998;273:19929-32.
- Magrane J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci 2005;25:10960-9.
- Zhou J, Schmid T, Frank R, Brune B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1α from pVHL-independent degradation. J Biol Chem 2004;279:13506-13.