
Crosstalk between signaling pathways of adrenoreceptors and signal transducers and activators of transcription 3 (STAT3) in heart1
Introduction
Adrenoreceptors (AR) play essential roles in various physiological and pathophysiological processes of the heart by means of subtype-specific signaling. Current clinical therapeutic strategies against this system are therefore an important part of the management of many disorders, such as coronary artery disease and chronic heart failure. While it is known that signal transducers and activators of transcription 3 (STAT3) as a cytoplasmic transcription factor mainly mediates cytokine- and growth factor-directed transcription under the stimulation of receptors, numerous data have shown that myocardial STAT3 activity is required for regulating various processes such as cardiac growth, function, tissue architecture (neovascularization and ventricular remodeling post-myocardial infarction), cellular survival[1], apoptosis, and even providing protection against various cardiovascular stresses (eg ischemia)[2]. For instance, targeted the disruption of the mouse STAT3 gene is associated with embryonic lethality[3]. Moreover, cardiomyocyte-restricted STAT3 knock-out mice exhibited greater inflammation, cardiac fibrosis, and heart failure with advanced age[4]. Endogenous STAT3 has recently been shown to improve cardiac function, increase vascularization, exhibit anti-apoptotic effects, and regenerate cardiomyocytes following stimulation with granulocyte colony stimulating factor in the infarcted mouse heart[5]. Interestingly, accumulating evidence suggests there is crosstalk between adrenoreceptors and the STAT3 signaling pathways. Elucidating the potential mechanisms of this crosstalk may provide new therapeutic targets for heart disorders. This article reviews the current understanding of these signaling pathways, and importantly details mechanisms of potential crosstalk.
Overview of STAT3 signaling
Intense investigation into STAT transcript factors has shown them to have important roles in tumor genesis, inflammation, and homeostasis[6]. Seven mammalian STAT proteins have been identified and designated as STAT1, 2, 3, 4, 5α, 5β, and 6. STAT3 ranges in size from 750 to 850 amino acids, contains 6 conserved domains (an amino terminal domain, a coiled-coil domain, a DNA-binding domain, a linker domain, a Src homology (SH) 2 domain, and a transcriptional activation domain), and exists in 3 natural isoforms (STAT3α, STAT3β, and STAT3γ). The C-terminal-truncated STAT3β and STAT3γ behave as dominant–negative proteins which functionally compete with their full-length counterparts to alter or inhibit target gene expression. Several studies have implicated STAT3 in regulating fundamental cellular biological processes such as proliferation, differentiation, malignant transformation, survival, and apoptosis[7].
New insights into the STAT3 signaling pathway The predominant receptors coupled to STAT3 signaling are the interleukin-6 (IL-6) receptor family, which has a gp130-associated signal transducer and interferon receptors. Although these receptors do not possess tyrosine protein kinase activity, their cytoplasmic domains contain binding sites of tyrosine kinases. Additionally, mechanical stretch also initiates the STAT3 pathway in rat cardiomyocytes. Upon ligand binding, the receptors undergo a conformational change in their cytoplasmic domain facilitating the recruitment of Janus kinases (JAK), which confer tyrosine kinase activity. JAK catalyze ligand-induced auto-phosphorylation and phosphorylation of tyrosine residues on the receptor, creating STAT3-docking sites, which leads to recruitment and phosphorylation of tyrosine residues on STAT3. Moreover, JAK also may generate a docking site for the SH2 domain containing other signaling molecules, including protein phosphatases and other adaptor proteins such as Shc, growth factor receptor-binding protein 2 (Grb2), Cbl, the p85 subunit of phosphatidyl-inositol-3 kinase (PI3K), therefore providing a platform for crosstalk between STAT3 signaling and other signal transduction pathways.
Alternatively, many growth factor receptors such as epidermal growth factor (EGF) can directly phosphorylate STAT3 through their intrinsic receptor tyrosine kinase activity. Accumulating evidence indicates that G-protein-coupled receptors (GPCR) (eg AR, angiotensin II receptors, and formyl peptide receptor like-1) can also induce phosphorylation of STAT3 at tyrosine residue 705 (STAT3-Tyr705) and serine residue 727 (STAT3-Ser727)[8]. Notably, some non-receptor tyrosine kinases such as the Src family (eg Src, Fyn, Lyn, and Lck) may also directly activate STAT3 in the absence of a classical stimulus[9]. In particular, there is evidence to indicate that cooperation between Src family kinases and JAK is required for full activation of STAT3, as inhibition of either kinase family suppresses STAT activity[10]. Also, Wen et al demonstrated that Bmx, one of 7 members of Bruton’s tyrosine kinase (Btk)/Tec non-receptor tyrosine kinase family, transactivated STAT-mediated gene expression in salivary and lung epithelial cells[11]. Important advances have been recently made with regard to the role of G proteins in regulating STAT3 signaling. Indeed, Src and STAT3 are underlying effectors of G proteins. In particular, Gαo and Gα2, which belong to the Gαi family of G proteins, induce transformation of fibroblasts through a Src/STAT3 signaling pathway. Phosphorylation of STAT3 facilitates homodimerization or heterodimerization through reciprocal SH2-phosphotyrosine interactions. STAT3 dimers are rapidly transported from the cytoplasm to the nucleus, and are dependent on the interactions of certain domains, such as coiled-coil and DNA-binding domains, with the nuclear pore complex involving importin-α3[12]. Cytoplasmic trafficking of STAT3 is an active process and the phosphorylation of STAT3-Tyr705 is not sufficient for the nuclear translocation of STAT3. STAT3 co-localizes with endocytic vesicles in transit from the cell membrane to the perinuclear region, in response to growth factor stimulation. The disruption of endocytosis, with specific inhibitors, blocks STAT3 nuclear translocation and STAT3-dependent gene regulation. These results suggest that cytoplasmic transport of STAT3 may be dependent on receptor-mediated endocytosis following receptor activation[13]. Once in the nucleus, it regulates transcriptional activity of target genes by binding to specific DNA response elements.
In contrast, 3 export signals have been defined in the C-terminal part of the coiled-coil domain, the DNA binding domain, and the linker domain (ie STAT3306–318, STAT3404–414, STAT3524–535), which play an important role in regulating nuclear export of STAT3[14]. Recently, STAT3 has been shown to bind to CREB-binding protein (CBP) and acetylated histone H4 within the nucleus to form dot-like structures, termed STAT3 nuclear bodies, that may be either directly involved in active gene transcription or serve as reservoirs of activated STAT3[15]. Furthermore, STAT3 may also interact with other transcription factors to modulate target gene transcrip-tion. For example, LIM homeodomain transcription factors such as islet1 (Isl1), which is important for the differentiation of motor neurons and organogenesis of the heart, interacts with JAK and STAT3 in COS-1, HepG2, and a human neuroblastoma cell line, SH-SY5Y, to form a complex. This complex then activates JAK1 by phosphorylation, which in turn increases tyrosine phosphorylation, facilitating STAT3 recruitment, leading to increases in DNA binding activity and expression of target genes[16]. Other transcription factors known to interact with STAT3 include c-Jun, Sp1, and nuclear factor-κβ (NF-κβ)[17].
Recent studies indicate that the formation of STAT dimers may occur independently of tyrosine phosphorylation and that substantial levels of STAT proteins occur in the nucleus of unstimulated cells. STAT3 may continuously shuttle between the cytosol and the nucleus of unstimulated cells in a manner independent of phosphorylation of Tyr705. Further-more, confocal real time imaging and pulse fluorescence localization after photobleaching technology indicated that the decrease in STAT3 nuclear export contributed to the nuclear accumulation of STAT3 following stimulation with IL-6[18]. Increasing evidence indicates that post-transcriptional modification may have an important role in the activation of STAT3. Yuan et al recently reported that STAT3 was also acetylated on lysine residue 685[19]. Correspondingly, unphosphorylated STAT3 (ie complete absence of tyrosine phosphorylation) can also drive gene expression, such that increases in STAT3 levels that follow its IL-6-dependent activation by tyrosine phosphorylation are reported to drive a distinct subset of genes. Furthermore, mRNA or proteins whose expression is driven by overexpression of unphosphorylated STAT3 have been presented at high levels in many cancers. Hence, STAT3 serves 2 quite distinct roles in cytokine-dependent transcription: (i) as a part of the primary response through the action of STAT3 dimers; and (ii) as a secondary part of the complete response through the action of increased amounts of unphosphorylated STAT3[20].
The role of STAT3 on transcription is similar with STAT1, which drives the constitutive expression of several genes in the complete absence of tyrosine phosphorylation, a function quite distinct from its role in inducible, phosphorylation-dependent gene expression in response to interferons and other cytokines. However, there is no data to indicate that STAT3 is methylated despite several studies showing that methylation of STAT1/6 is essential for their activation. STAT3 is phosphorylated by many different kinases which would appear to be dependent on species, cell type, and eliciting stimulus. For example, Ser727 is phosphorylated by several kinases, including Cdk5, H7-sensitive kinase, protein Kinase Cδ, zipper-interacting protein kinase[21], PI3K, and mitogen-activated protein kinase (MAPK) cascade, that is, MEK kinase 1, SEK-1/MKK-4, extracellular signal-regulated kinase (ERK), p38, c-Jun NH2-terminal kinase (JNK), ribosomal protein S6 kinases, mitogen- and stress-activated protein kinase 1, transforming growth factor β-activated kinase 1[22], and Ca2+/calmodulin-dependent kinases. Indeed, signal transduction from the plasma membrane to the nucleus by STAT proteins is widely represented as exclusively a soluble cytosolic process. Shah et al recently showed that the endocytic pathway displayed important effects in IL-6/STAT3 signaling[23]. For example, IL-6 could enhance the association of cytoplasmic phosphorylated STAT3-Tyr705 with the purified early endosome fraction. Also, the inhibition of endocytosis by transfection with dominant negative amphiphysin A1, epsin 2a, dynamin K44A, and the clathrin light chain, resulted in an inhibition of IL-6-stimulated STAT3 transcriptional function, indicating that endosome-mediated trafficking of STAT3 may be required for optimal signal trans-duction.
Downregulation of STAT3 signaling At least 8 different mechanisms have been implicated in downregulating STAT3 signaling. However, the molecular basis of these has not been well characterized. Several cytoplasmic tyrosine phosphatases, including tyrosine phosphatases containing a SH2 domain (SHP-1), CD45, and protein tyrosine phosphatase 1B, are implicated in the dephosphorylation of STAT3 signaling. In addition, suppressors of cytokine signal proteins (SOCS) are potentially key negative regulatory modulators of STAT3 signaling as they bind to receptor sites and/or JAK catalytic sites. Moreover, the protein inhibitor of activated STAT3 (PIAS3) was found to be specific for the inhibition of activated STAT3[24]. Scaffolding proteins such as the STAT3-interacting protein (StIP1), a novel protein consisting of 12 WD40 repeats, can also inhibit STAT3 signaling by modulating the formation of multiprotein complexes that are central in the regulation of signal transduction, transcription, and targeted proteolysis. Collum et al demonstrated that StIP1 had a high affinity for unphosphorylated JAK and STAT3, and when overexpressed blocks STAT3 activation and dimerization/DNA binding, nuclear translo-cation, and reporter gene transcription following stimulation with IL-6[25].
Other proteins involved in the regulation of STAT3 include genes associated with retinoid-interferon-induced mortality-19, which regulates nuclear translocation of STAT3[26], and TEL/ETV6, a member of the ETS family of transcription factors that represses STAT3 transcriptional activity independent of TEL DNA binding[27]. A Ras homologue member I (ARHI), a novel imprinted tumor suppressor gene, recently was shown to form a complex with STAT3 in the cytoplasm which resulted in the prevention of STAT3 accumulation in the nucleus following stimulation with IL-6. ARHI markedly reduced STAT3 binding to DNA and STAT3-dependent promoter activity while only moderately affecting STAT3 phosphorylation[28]. In addition, the ubiquitin-proteasome degradation pathway, which modulates the turnover of cytokine receptors and activated JAK through various E3 ligase com-plexes, may also serve as a highly specific mechanism to inhibit STAT signaling[29]. Finally, receptor internalization and secretion of soluble receptors have also been implicated in the regulation of the JAK/STAT3 pathway.
In light of this, it is clear that the regulation of STAT3 signaling is a complex process involving a network of partners including membrane receptors, interaction with membrane-proximal region signaling molecules, modulation of STAT3 through protein degradation/phosphorylation/acetylation, and crosstalk with the other signal transduction pathways, in order to facilitate intra-nuclear coordination with the other transcription factors.
Overview of AR signaling in the heart
As AR belong to GPCR family, they utilize various G-protein subunits as part of their signaling cascade. Cardio-myocytes express at least 6 AR subtypes, including α1A, α1B, α1D, β1, β2, and β3. With respect to the classic pathway of AR signaling, it is accepted that α-AR couple Gq/phospholipase Cβ (PLCβ)/inositol triphosphate (IP3) and activate diacylglycerol/calcium and protein kinase C (PKC) signaling, while β-AR are coupled to the Gs/i/adenylate cyclase/cAMP/protein kinase A (PKA) pathway. On the other hand, increasing evidence suggest novel mechanisms through which AR can also induce responses by activating transduction systems that do not involve G proteins through the receptor–protein interaction. For example, Shenoy et al recently demonstrated that β-arrestins mediated β2-AR signaling to ERK1/2 independent of G-protein activation in HEK293 cells[30]. However, the significance of this novel signaling mechanism in the cardiovascular system remains unclear.
α1-AR signaling α1-AR signaling in the heart is diverse and involves the activation of multiple signaling pathways. It is well established that 3 subtypes of α1-AR (α1A, α1B, and α1D) mobilize intracellular Ca2+ and activate PKC via Gq/11. However, α1-AR have also been shown to activate pertussis toxin-sensitive G proteins, such as Go, and mediate the contractile response in rat aorta. In addition, several lines of evidence indicate that α1-AR are also coupled to alternative effectors, such as phospholipase D, calcium/calmodulin sensitive kinases, the Na+/H+ exchanger, Na+, K+-ATPase, and various ion currents including the L-type Ca2+ current, the transient outward current, the delayed rectifier K+ current, and the acetylcholine-activated K+ current. Thus, α1-AR underpin many key processes in the heart including myocardial contraction, growth response of cardiomyocytes/fibroblasts, the cell cycle, differentiation and hypertrophy, as well as survival and apoptosis[31]. The function of α1-AR in the cardiovascular system has been extensively studied[32]. Moreover, studies in recent years utilizing transgenic mice overexpressing α1-AR, or with targeted disruption of genes associated with α1-AR signaling in the myocardium, have provided insights into mechanisms of α1-AR-induced cardiac hypertrophy. Cardiac overexpression of Gq in transgenic mice results in hypertrophy, decreased ventricular function, and loss of α-AR inotropic responsiveness. In contrast, upon overexpressing a carboxyl-terminal peptide of the α subunit Gαq (a functional Gq knockout), pressure overload induced myocardial hypertrophy was reduced by 60%–70%, which implies that 30%–40% of the hypertrophic response is independent of PLC/PKC[33]. O’Connell et al recently reported that gene ablation of the α1A and α1B subtypes in mice resulted in a maladaptive form of reactive cardiac hypertrophy from pressure overload, with a predisposition to heart failure[34]. Given the potentially important role of α1-AR signaling in the heart, further work is necessary to elucidate crosstalk between this pathway and others involved in cardiac hypertrophy signaling. Indeed, recent studies from our laboratory have shown that filamin C can interact with 3 subtypes of α1-AR[35], while bone morphogenetic protein-1 can only specifically interact with α1A-AR. Another protein that interacts with α1-AR is spinophilin[36].
β-AR signaling Within the heart, 3 subtypes of β-AR have been cloned and identified pharmacologically. Recent pharmacological studies have identified the existence of a fourth subtype of β-AR in human and rat cardiac tissue, which remains to be cloned[31]. β-AR are associated with myocardial contraction, growth control, cell survival, and apoptosis. Several β-AR subtypes can initiate multiple intracellular signaling pathways in addition to Gs/adenylate cyclase/PKA. For instance, recent studies demonstrate that β2-AR are coupled to MAPK and cytoplasmic phospholipase A2, via Gi in cardiomyocytes, which enhance cardiac contraction and increase cardiomyocyte survival via Gi/PI3K/Akt[37] and proliferation during early postnatal life. Wang et al recently reported that in the PLC epsilon-deficient mouse, cardiac contractility was attenuated in response to isoprenaline and was independent of both β-AR-mediated cAMP production and the size of the sarcoplasmic reticulum calcium pool[38]. Furthermore, these knock-out mice were more susceptible to the development of hypertrophy than wild type mice. Hence, β-AR are believed to utilize different signaling molecules to activate alternative compensatory mechanisms under conditions of altered or defective classic signaling.
Relatively little is known about the precise signaling pathway associated with the stimulation of β3-AR in the myo-cardium. Until recently, β3-AR were purported to be functional via the activation of adenyl cyclase and cAMP-dependent phosphorylation. However, β3-AR do not possess a C-terminal phosphorylation site for PKA or the β-AR kinase; therefore, its signaling pathway may be different from that of β1- and β2-AR. In human ventricular biopsies and β3-AR knock-out mice, β3-AR was associated with reduced cardiac contractility through Gi/o and production of nitric oxide via endothelial constitutive nitric oxide synthase[39]. In contrast, overexpression of β3-AR in myocardium increases adenyl cyclase activity and enhances cardiac performance Interes-tingly, it appears that a reciprocal relationship exists between β3- and β1-AR in cardiomyocytes. Germack et al recently observed that β3-AR were functionally upregulated and coupled to the Gi protein in rat neonatal cardiomyocytes following chronic exposure to noradrenaline, while β1- and β2-AR were downregulated[40]. This pattern is also observed in human heart failure, as downregulation of β1-AR is accompanied by a 2–3-fold upregulation of β3-AR expression[41]. Similar to α1-AR signaling, β-AR are differentially associated with a variety of proteins other than G proteins[42]; those which interact with β1-AR include endophilin-1, PSD95, cyclic nucleotide Ras guanine nucleotide exchange factor (associated with Ras activation), and GAIP-interacting protein carboxyl terminus. Likewise, β2-AR have been shown to interact with proteins, such as the adaptor protein, Grb2, and a non-receptor tyrosine kinase, Src. In addition, β-AR signaling is modulated through the coordinated actions of various cytoplasmic enzymes including G-protein-coupled receptor kinases, which phosphorylate and thus desensitize the receptor, cyclic nucleotide phosphodiesterases which hydrolyze cAMP, and phosphatases which dephosphorylate phosphoproteins[31]. Differential expression patterns, heterogeneity of tissue distribution, and intracellular compartmentalization of these proteins may contribute to diversity in downstream signaling of β-AR. Therefore, the possibility exists for crosstalk among the various signaling pathways and regulation of a multitude of biological processes.
Crosstalk between AR signaling and the STAT3 pathway
Numerous studies have investigated the role of STAT3 and AR (α1 and β) in cardiac remodeling/hypertrophy, ischemic injury, and ischemic preconditioning in recent decades. Interestingly, accumulating evidence indicates that these signaling pathways undergo reciprocal crosstalk[43]. For example, we have recently demonstrated that STAT3 plays a crucial role in mediating cardiac hypertrophy, induced by α1-AR stimulation in neonatal rat cardiomyocytes (unpublished data), although the underlying mechanisms are not fully understood.
Intercellular crosstalk: interaction between cardio-myocytes and non-cardiomyocytes Several studies show that interaction between cardiomyocytes and non-cardiomyo-cytes may play a role in mediating crosstalk between AR and STAT3 signaling (Figure 1). We have previously demonstrated that isoprenaline per se does not induce the phosphorylation of STAT3-Tyr705 in mouse cultured ventricular cardiomyocytes or fibroblasts. In contrast, in vivo administration of isoprenaline (ip 15 mg/kg) caused delayed phosphorylation of STAT3-Tyr705 in myocardium at 60 and 120 min, which was accompanied by an increase in myocardial expression and serum levels of the IL-6 family (mainly IL-6); this activation of STAT3 was abolished by phosphodiesterase inhibition[44], suggesting that the delayed activation might be mediated by cAMP. Interestingly, using anti-murine IL-6-neutralizing antibody could significantly inhibit isoprenaline-induced phosphorylation of STAT3-Tyr705, suggesting that IL-6 might mediate isoprenaline-induced STAT3 activa-tion. In vitro studies further show that the secretion of members of the IL-6 cytokine family induced by isoprenaline was from mice fibroblasts rather than cardiomyocytes[44]. Moreover, evidence supporting a paracrine mechanism was identified by Fredj et al, using an in vitro co-culture of cardio-myocytes and fibroblast, where cardiomyocyte hypertrophy was found to be dependent on the secretion of angiotension II from fibroblasts[45]. More recently, our data demonstrate that isoprenaline-induced secretion of IL-6 in cardiac fibroblasts from mice is mainly mediated by the α2-AR-Gs-AC-cAMP (but not PKA) signaling cascade, whereas inhibiting the Gi/PI3K pathway significantly enhanced, while pre-treatment with SB203580 (an effective blocker of p38-MAPK) abolished, the induction of IL-6 by isoprenaline. These data indicate that the mechanism mediating crosstalk between these 2 pathways may centre on IL-6-mediated phosphorylation of STAT3[46]. Indeed, it is clear that communication between cardiomyocytes and fibroblasts, which are major components of myocardium, appears to be more complex than initially anticipated.
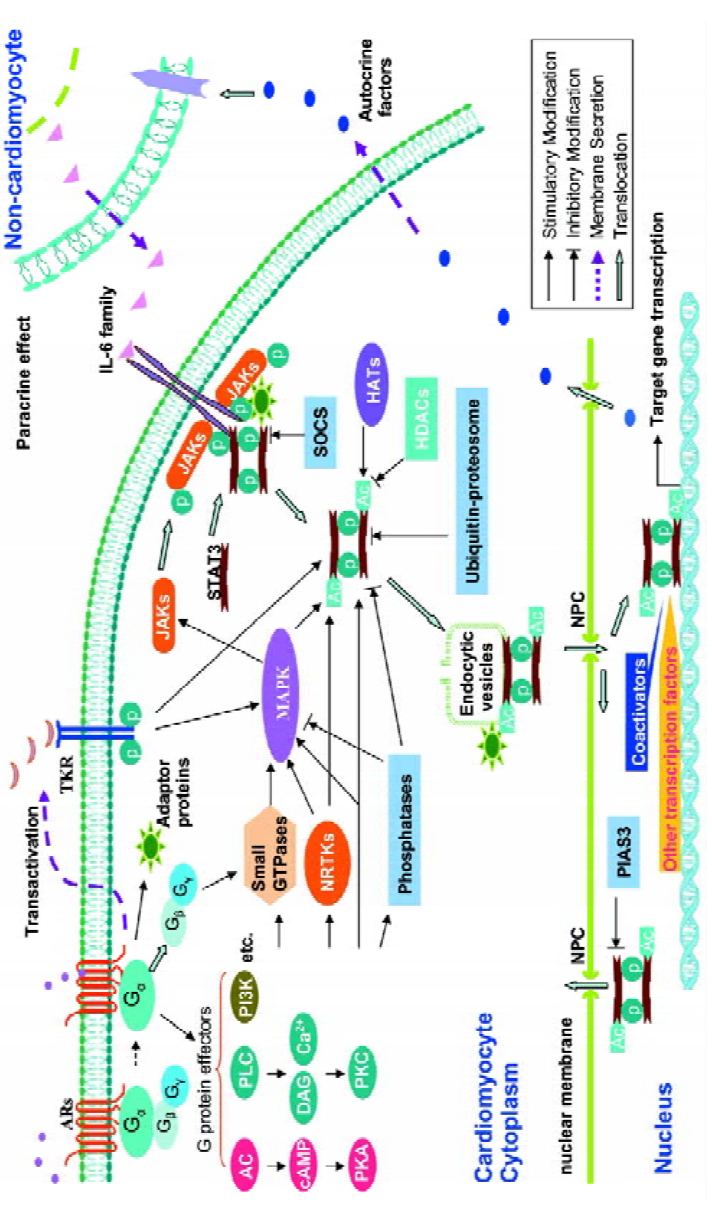
Intracellular crosstalk Sasaguri et al were the first to report that α1B-AR stimulation caused phosphorylation of tyrosine residues in JAK2 and STAT1[47]. In particular, they showed that in vascular smooth muscle cells, phenylephrine-induced protein synthesis was dependent on STAT1 activation and could be inhibited by AG490, an inhibitor of JAK2. Furthermore, Briest et al demonstrated that intravenous administration of noradrenaline in rats caused phosphorylation of STAT3-Tyr705 in cardiomyocytes, but not non-myocytes, but was accompanied by an increase in IL-6 mRNA levels in both cardiomyocytes and non-cardiomyocytes. However, IL-6 and STAT3 levels were unchanged in non-cardiomyocytes following noradrenaline infusion, despite phosphorylation of STAT3-Try705 being downregulated, which was in keeping with in vitro results. Systematic analysis of NF-IL-6, IL-6, IL-6 receptor, and STAT3 mRNA expression in myocardium following noradrenaline administration supports the hypothesis that noradrenaline-induced cardiac hypertrophy may involve sequential signaling via NF-IL-6, IL-6, IL-6 receptors, and STAT3[48]. Similarly, our laboratory has recently shown that in cultured rat cardiomyocytes α1-AR stimulation causes phosphorylation of STAT3 at Ser727, as early as 5 min and delayed phosphorylation of Try705. Notably, cardiac hypertrophy in response to α1-AR activation can be significantly relieved via AG490 or an inhibitory peptide of STAT3 (unpublished data). However, the precise mechanisms remain to be elucidated.
Mechanisms underlying AR-mediated activation of STAT3 signaling
Transactivation In recent years, increasing evidence suggests that transactivation of receptor tyrosine kinase by GPCR is a general phenomenon. Stimulation of many GPCR by a diverse range of agonists including lysophosphatidic acid, angiotensin II, endothelin, and bradykinin, are able to activate receptor tyrosine kinase by transactivation[49]. For example, lysophosphatidic acid induces the release of the heparin-binding EGF by a metalloproteinase, thereby resulting in the transactivation of EGF receptors[50]. In addition, GPCR have also been shown to transactivate other receptor tyrosine kinases, such as the vascular endothelium growth factor, the platelet-derived growth factor, and the insulin-like growth factor, resulting in the formation of receptor tyrosine kinase/GPCR complexes that trigger a complex sequence of intracellular signaling events which include STAT3 signaling.
AR are also capable of transactivating receptor tyrosine kinases (Figure 1). For instance, in neonatal rat cardiomyocytes, binding of phenylephrine to α1-AR results in EGF receptor transactivation, in turn regulating transcription of atrial natriuretic peptide by an ERK-mediated pathway[51]. Similarly, β2-AR-mediated transactivation of the EGF receptor induces proliferation of cardiac fibroblasts through a P13K/ERK pathway[52]. Intriguingly, we recently demonstrated that α1-AR-induced phosphorylation of STAT3–Tyr705 required transactivation of EGF receptors (unpublished data). Although investigations of transactivation of GPCR and receptor tyrosine kinase have provided new insights into elucidating the interactions between various signal transduction pathways, the precise mechanisms remain unclear. Furthermore, it is important to be able to differentiate between a receptor-complex-mediated signaling pathway and simple receptor tyrosine kinase-mediated signaling.
G proteins Recent reports indicate that Gαs and Gαi, which are important subunits of G-protein-mediated β-AR signaling, directly modulate Src kinase activity by binding to its catalytic domain, which induces a conformational change enhancing accessibility to its active site[53]. These results suggest that other members of the Src tyrosine kinase family may also be downstream effectors of G-protein signaling. A Src/STAT3 pathway has been established in various cells types. Previous studies indicate that β2-AR stimulation is associated with phosphorylation of its C-terminal (Tyr350), which exposes a binding site for the SH2 moiety of Src. The resulting binding between β2-AR and Src leads to the phosphorylation of Src and the activation of a G-protein-linked receptor kinase 2[54], which in turn phosphorylates Ser/Thr residues at the C-terminal of the β2-AR, prompting the binding of β-arrestin and internalization of the receptor –– a critical step in the resensitization and recycling of the receptor[55]. In addition to the Src/STAT3 pathway, several lines of evidence show that Gαs and Gαi also participate in the activation of MAPK signaling. Therefore, the possibility exists for Gαs and Gαi to modulate STAT3 activation via a MAPK/STAT3 pathway (as discussed later). In addition, Gq and Gi have also been shown to activate members of the Btk family[56]. While Btk are associated with MAPK signaling, some members of the family, such as Bmx can directly induce STAT activation[11]. Although all 3 α1-AR subtypes are frequently coupled to PLC-β activation, via Gq/11, α1B-AR can interact with Gα14 and Gα16, while α1D-AR couple to Gα14 rather than Gα16[57]. Ho et al report that Gα14 is associated with both α2- and β2-AR signaling[58]. In HEK293 cells, STAT3 activation is mediated through Gα14 and involves multiple intermediates including PLC-β, calmodulin-dependent kinase II, PKCα,ε, c-Src, JAK2/3, Ras/Rac1, Raf-1, and ERK[59]. Likewise, Gα16 also activates STAT3 via a c-Src/JAK- and ERK-dependent cascade[60]. However, at present there are no data available to support the existence of Gα14 in the heart. In addition to Gα, other G-protein subunits (Gβ,γ) also modulate the activity of Btk through direct binding to their PH/TH domain[61]. This raises the possibility that Gβ,γ could also mediate Btk/STAT3 signaling.
cAMP/PKA pathway cAMP/PKA signaling is the prototypical pathway associated with β-AR stimulation and has been implicated in a vast array of cellular processes including cell proliferation and contraction. However, the response would appear to be dependent on cell type and nature of the stimulus. New evidence indicates that increased cAMP levels inhibit angiotensin II-mediated JAK/STAT3 activation by a variety of mechanisms. Giasson et al observed that in vascular smooth muscle cells, increased cAMP abolished Tyk2 phosphorylation and inhibited JAK-attenuated protein synthesis and vasoconstriction following exposure to angiotensin II[62]. Interestingly, cAMP-elevating agents were also shown to inhibit IL-6-induced STAT activation in monocytes[63]. Together, these data establish a link for crosstalk between cAMP and JAK/STAT3. At present, it is not known how cAMP inhibits Tyk2 activation. However, it may involve effects on PKA since pharmacological inhibition of PKA abolishes the effects of cAMP. Tyk2 may be a downstream target of PKA. Alternatively, PKA may directly or indirectly activate protein tyrosine phosphatases, such as SHP-1, resulting in the dephosphorylation of JAK. In contrast, Park et al found that an increase in cAMP was associated with the activation of STAT3, and inhibiting PKA did not alter STAT3 activation in FRTL-5 thyrocytes[64]. The picture may be even more complex as the inhibition of PKC also blocked cAMP-induced activation of STAT3. These results suggest that the connection between the cAMP and JAK/STAT3 pathway is diverse and dependent on both cell type and species studied.
Small GTPase Small GTPases are a family of more than 100 monomeric G proteins which act as molecular switches in signaling cascades and are modulated by guanine nucleotide exchange factors. They can be divided into 6 subfa-milies, Ras, Rho, Rab, Arf, Ran, and Rad. Ras and Rho are known to play important roles in gene regulation and cytoskeleton assembly, while the other subfamilies are primarily involved in cytoplasmic transportation and assembly of the microtubule spindle. Many studies indicate that small GTPases are involved in the activation of STAT3. Simon et al demonstrated that STAT3 formed a complex with activated Rac1 (a member of the Rho subfamily), which increases Ser727 phosphorylation of STAT3. In support of this, overexpres-sion of a dominant negative mutant of Rac1 inhibited EGF-mediated activation of STAT3[65]. Pelletier et al subsequently showed that Rac1 was necessary for angiotensin II-induced activation of JAK and involved the generation of reactive oxygen species via NADPH. Interestingly, this caused a biphasic response with regard to phosphorylation of STAT3-Tyr705. The initial phase associated with JAK activation had a quick onset, reaching its peak within 3–6 min, before returning to baseline by 15 min. In the second or delayed phase, JAK activation occurred at 60 min and was maintained for a further 60 min. As the delayed phase was abrogated by actinomycin D (transcription inhibitor), it suggests that the delayed activation phase is dependent on de novo protein synthesis, possibly involving IL-6[66]. Similarly, other members of the Rho subfamily, namely Rac and Cdc42, are implicated in β2-AR-mediated activation of JNK in smooth muscle cells. Furthermore, β-AR-induced apoptosis would appear to involve the activation of Rac1 in rat cardiomyo-cytes[67]. Although members of the Rho family have a role in α1-AR-mediated myocardial hypertrophy, it remains to be established whether small GTPases, such as Rho, link AR to STAT3 signaling in the heart.
Scaffold and adaptor proteins Scaffolding and adaptor proteins are emerging as “bridges” which organize the spatial orientation of signal transduction pathways within mammalian cells. For example, growth factor receptor-binding protein 2 (Grb2)-associated binder-1 (Gab1), one of 3 members of the Gab scaffolding protein family, interacts with various signal molecules associated with the growth factor receptor, the cytokine receptor, and GPCR signaling, such as protein tyrosine phosphatase SHP2, the p85 subunit of PI3K, PLCγ, and Grb2. In cultured rat cardiomyocytes, stimulation with leukemia inhibitory factor (LIF) induces phosphorylation of Gab1, and initiates its interaction with SHP2 and p85. This Gab1-SHP2 interaction is crucial in mediating gp130-dependent cytokine-induced hypertrophy in cardio-myocytes and involves the activation of ERK5[68]. Interes-tingly, transient expression of Grb2 inhibits EGF-mediated STAT3 activation, while silencing Grb2 with RNA interference enhances STAT3 activation[69]. In addition, the direct binding of Grb2 to β2-AR mediates receptor internalization following stimulation with insulin[70]. Due to the number of reports documenting the interaction between scaffold proteins/adaptor proteins and AR or JAK/STAT3 signaling, it is not unreasonable to suggest that they may mediate crosstalk between AR signaling and STAT3 signaling through the formation of the signal molecules complex.
PKC PKC is generally activated in response to stimulation of GPCR and protein tyrosine kinase receptors, or via a non-receptor tyrosine kinase mechanism. It has been implicated in the regulation of cell differentiation, proliferation, apoptosis, and growth response. At least 11 isoforms of PKC have been identified and are classified into 3 major categories based on differential requirements for Ca2+ and lipids. Their downstream targets include the Rho kinase, ERK, calcium/calmodulin-dependent protein kinase II, and several ion channels. There is a considerable body of evidence to indicate that PKC participates in the activation of STAT3 in various cells, such as FRTL-5 thyrocytes, HepG2 cells, and monocytes, in response to receptor stimulation or mechanical stretch. For example, Schuringa et al demonstrated that in unstimulated HepG2 cells, PKCδ strongly associated with SEK-1/MKK-4 and was released from this complex in an IL-6-dependent manner to phosphorylate STAT3-Ser727, but not STAT3-Tyr705[71]. This occurs independently of JNK-1. Rottlerin, a PKCδ inhibitor, causes a dose-dependent reduction in STAT3 transactivation and is coupled to a decrease in IL-6-induced STAT3-Ser727 phosphorylation; IL-6-induced STAT3-Tyr705 phosphorylation is unaffected. Similarly, overexpression of dominant negative PKCδ also reduces IL-6-induced phosphorylation of STAT3-Ser727, but not STAT3-Tyr705. Several recent reports have shown a direct association between PKC and STAT in the heart. Wang et al demonstrated that PKC phosphorylation enhanced transcription factor GATA-4 DNA binding activity and STAT1 interacted with GATA-4 to synergistically activate angiotensin II and growth factor-inducible promoters[72]. Xuan et al reported that ischemic preconditioning was associated with simultaneous activation of STAT1 and STAT3 via 2 parallel signal transduction pathways, JAK1/2/STAT and a PKCε/Raf-1/MEK1/2/ERK cascade[73]. Our recent studies indicate that chelerythrine, an inhibitor of PKC, can inhibit the delayed phosphorylation of STAT3 induced by α1-AR stimulation in rat cardiomyocytes. Consistently, PMA, via the activation of PKC, also can evoke the phosphorylation of STAT3-Tyr705 (unpublished data). However, it is unclear which subtype of PKC is responsible for the induction of STAT3 activation by α1-AR. In addition, it is noteworthy that PKC can phosphorylate its substrates on serine residues, thus, per se may not directly activate STAT3 by tyrosine phosphorylation. Novotny-Diermayr et al provided some evidence to partly explain this puzzle[74]. As the catalytic domain of PKCδ interacts with the SH2 domain and part of the adjacent C-terminal transactivation domain of STAT3, this interaction does not seem to depend upon phosphotyrosine SH2-mediated binding. However, it significantly enhances the interaction of STAT3 and the IL-6 receptor subunit, gp130, facilitating STAT3 phosphorylation by gp130 receptor stimulation.
Cytoplasmic protein tyrosine kinase As mentioned earlier, STAT3 is also activated by certain non-receptor tyrosine kinases other than JAK. In particular, Src plays an important role in mediating STAT3 signaling. Src phosphorylates the adapter protein Shc, which in turn activates the Ras/Raf/ERK signaling cascade. It has been demonstrated that IL-3 stimulates c-Src kinase activity, which facilitates the binding of c-Src to STAT3, leading to the phosphorylation of STAT3 and its translocation to the nucleus. While a dominant negative mutant of JAK2 had no effect on IL-3-mediated activation of STAT3, a dominant negative mutant of c-Src abrogated STAT3 activation and inhibited IL-3-mediated myeloid cell proliferation. These results indicate that JAK and STAT phosphorylation events are mediated by 2 independent pathways[75]. Interestingly, many GPCR are able to increase the activity of Src tyrosine kinases through an ill-defined mechanism. Furthermore, various G-protein-mediated physiological functions are sensitive to tyrosine kinase inhibitors. In Src-deficient mouse embryonic fibroblasts, the internalization of β2-AR is prevented; however, they are still coupled to MAPK signaling[76]. It has been shown that Gαs and Gαi directly stimulate the kinase activity of downregul-ated Src and modulate Hck, another member of the Src family of tyrosine kinases, in a similar manner. There is evidence that small GTPases, such as Rap1 or Ral, may be involved in the mechanism by which Gαo activates Src. G proteins may also activate signaling molecules, such as Rap1, p38, and JNK, which go on to form a complex with Src resulting in the activation of Src. These results would indicate that direct modulation of Src kinases by G proteins may be a common phenomenon.
AR are associated with the activation of Src in various cells, including airway smooth muscle cells. For instance, β2-AR stimulation initiates the assembly of a protein complex containing the receptor-activated c-Src and β-arrestin[55]. Furthermore, it is generally accepted that Ras-dependent activation of MAPK by GPCR, requires the activation of the Src family tyrosine kinases. In cardiomyocytes, a Gi/Gβγ, Src, Ras/Raf-1, and ERK cascade mediate isoprenaline-induced myocardial hypertrophy[77]. In cardiac fibroblasts, pre-incubation with PP2 (Src inhibitor) decreases isoprenaline-mediated phosphorylation of EGF receptors and ERK activa-tion[52]. In PC12 cells stably expressing α1-AR, noradrenaline stimulates Src tyrosine phosphorylation, while PP2 completely blocked ERK activation and cell differentiation[78]. However, it remains to be established if there is a direct association among AR/Src/STAT3 in the heart.
MAPK MAPK represent a family of serine/threonine protein kinases and at least include ERK1/2, p38, JNK, and ERK5. They are involved in the activation of other protein kinases and transcription factors. MAPK share many upstream and downstream kinases and transcription factors that interact and integrate within these pathways. Several studies have indicated that GPCR activate MAPK cascades by multiple pathways, which include signaling molecules such as Ras, Raf, PKC, Ca2+, and even tyrosine kinase-dependent and independent pathways. AR-induced MAPK signaling has been intensively studied in various species and cell types (Figure 1). However, crosstalk between MAPK and JAK/STAT3 signaling would appear to be more complex and dependent on the species studied, cell type and nature of the stimulus. For example, IL-6 activates the Ras/MAPK cascade via JAK2, but MAPK also stimulate JAK phosphorylation[79].
Kunisada et al demonstrated that LIF caused tyrosine phosphorylation of gp130, JAK1, and STAT3[80]. They also demonstrated that the activation of gp130 could trigger, in parallel, both a MAPK cascade and STAT3 signaling in rat cardiomyocytes. IL-1β and LIF are potent stimuli which can cause cardiac hypertrophy in rat. Data indicate that IL-1β-induced phosphorylation of STAT3-Tyr705 occurred relatively late at 60 min, compared with that induced by LIF within 10 min. Interestingly, they were both associated with ERK activation. Pharmacological inhibition of ERK and p38 abolished the delayed phosphorylation of STAT3 and reduced atrial natriuretic factor expression by 70% following exposure to IL-1β[81]. In addition, angiotensin II induced rapid phosphorylation of STAT3-Ser727 in rat cardiomyo-cytes, which was accompanied by an initial dephosphorylation of STAT3-Tyr705 in the first 30 min and phosphorylation of STAT3-Tyr705 after 90 min. All of these effects were abolished by the ERK inhibitor, PD98059[82]. These data suggest an important role for ERK and p38 in the delayed activation of STAT3 induced by angiotensin II. Nevertheless, we recently demonstrated that, compared with JNK, ERK and p38 do not participate in α1-AR-mediated phosphorylation of STAT3-Tyr705. In contrast, the Gq/PLC/ERK pathway is important in the phosphorylation of STAT3-Ser727 induced by α1-AR stimulation in neonatal rat cardiomyocytes (unpublished data).
Interestingly, ERK-induced phosphorylation of STAT3-Ser727 is sometimes associated with the inhibition of STAT3-Tyr705 phosphorylation. Previously, studies in bone marrow cells, hepatocytes, and HEK293 cells indicated that MEK/ERK activation could rapidly (within 5 min) inhibit STAT3 tyrosine phosphorylation and DNA-binding activity following exposure to IL-6[83]. In contrast to this, phenylephrine inhibited IL-6-induced phosphorylation of STAT3, in primary culture hepatocyte and HepG2 cells, which overexpress α1B-AR via an ERK-dependent mechanism. This inhibition was relatively late (occurring 2 h post stimulation) and was abolished by either a tyrosine phosphatase blocker or MEK inhibitor, but not a JAK1/2 inhibitor[84]. However, this inhibitory mechanism requires further exploration. In contrast, recent data show Ser727 phosphorylation is required to achieve maximal transcriptional activity of STAT3. For example, Schuringa et al found that IL-6-induced transactiva-tion of STAT3 and Ser727 phosphorylation was independent of ERK-1 or JNK activity, but involves a signaling pathway that includes Vav, Rac-1, MEK kinase, and SEK-1/MKK-4[85]. The mechanism underlying the enhanced transcriptional activity of STAT3-Ser727 might involve the selective recruitment of co-activators, such as p300. While the AR, MAPK, and JAK/STAT3 signaling pathway are 3 important mechanisms mediating cardiac hypertrophy, elucidation of their interaction will provide new insights for the development of novel clinical strategies for this disorder.
Acetylation of STAT3 It is generally accepted that the phosphorylation of STAT3-Tyr705 plays a critical role in dimerization and induction of gene transcription, and that post-translational modification of Ser727 modulates STAT3 functionality. Nevertheless, accumulating evidence indicates that unphosphorylated or tyrosine-mutated STAT proteins can still form dimers and induce gene transcription[86]. Therefore, another type of regulation must contribute to the stable dimerization of STAT. Recently, Wang et al showed that STAT3 could be acetylated in the C-terminal transcriptional activation domain at lysine 685 by its co-activator, p300/CBP, both in vivo and in vitro[87]. This modification enhances its sequence-specific DNA binding ability and transactivation activity. In contrast, histone deacetylase (HDAC) acts as a negative modulator. Around the same time, Yuan et al[19] also reported the same post-translational modification in other cells following cytokine stimulation. In PC3 cells co-transfected with p300 and a series of STAT3 mutants (Tyr705, Ser727, and Arg585), STAT3 was still acetylated, indicating that acetylation occurs independently of phosphorylation and SH2 domain activity. Moreover, the acetylation of Lys685 plays a critical role in STAT3 dimeriza-tion, transcription of cell growth-related genes, and modulation of the cell cycle in response to cytokines. Although STAT3 deacetylation is mediated by type 1 HDAC (mainly HDAC3), there is still controversy over which lysine residues in STAT3 are acetylated. Based on previous studies showing that the IL-6/JAK/STAT3 pathway is involved in the transcriptional modulation of human angiotensinogen (hAGT) in hepatocytes, Ray et al recently demonstrated that STAT3 was acetylated at its NH2 terminus on lysine residues 49 and 87[88]. A double-point mutation of STAT3 at these 2 conserved sites caused a 3-fold decrease of IL-6-induced hAGT transactivation. Furthermore, binding of HDAC1 and 4 (particularly HDAC1) to activate STAT3 acts as an intra-nuclear molecular switch. This controls the duration of the STAT3 transcriptional response and contributes to the replenishment of cytoplasmic pools depleted of latent STAT3. Therefore, further work is required to investigate the cell-type-specific differences in acetylation sites of STAT3. A considerable body of data indicates that activation of ERK is a key factor in mediating phenylephrine-induced cardiac hypertrophy. Interestingly, phenylephrine could increase the transcriptional activation ability of CBP and p300, as this effect is dependent upon its ability to activate ERK signaling[89]. More importantly, the inhibition of CBP and p300 decreased atrial natriuretic factor gene expression following exposure to phenylephrine. Based on this and the fact that CBP and p300 modulate STAT3 acetylation, whether it is possible that a relationship exists between AR/ERK/CBP/p300 and STAT3 remains to be established along with its biological significance.
Summary
There is mounting evidence of the crucial roles of AR and STAT3 signaling pathways in various heart pathophysiological processes. Data suggest that the complex crosstalk between the 2 signaling pathways appears to depend on species/tissue type, development status, and stimulus. Elucidation of the potential mechanisms of their crosstalk, occurring at multiple cascade levels by different manners, will provide new targets for the development of novel clinical strategies for heart disorders.
Acknowledgements
We thank Dr James P SPIERS and Dr Elizabeth J KELSO for valuable revision on the manuscript.
References
- Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr, et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 1999;97:189-98.
- Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther 2005;107:131-7.
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse STAT3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997;94:3801-4.
- Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA 2003;100:12929-34.
- Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-STAT pathway in cardiomyocytes. Nat Med 2005;11:305-11.
- Calò V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, et al. STAT proteins: From normal control of cellular events to tumorigenesis. J Cell Physiol 2003;197:157-68.
- Levy DE, Lee C. What does STAT3 do? J Clin Invest 2002;109:1143-8.
- Kisseleva T, Bhattcharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002;285:1-24.
- Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, et al. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol 2005;25:4826-40.
- Ingley E, Klinken SP. Cross-regulation of JAK and Src kinases. Growth Factors 2006;24:89-95.
- Wen X, Lin HH, Shih HM, Kung HJ, Ann DK. Kinase activation of the non-receptor tyrosine kinase Etk/BMX alone is sufficient to transactivate STAT-mediated gene expression in salivary and lung epithelial cells. J Biol Chem 1999;274:38204-10.
- Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc Natl Acad Sci USA 2005;102:8150-55.
- Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J 2002;21:3255-63.
- Bhattacharya S, Schindler C. Regulation of Stat3 nuclear export. J Clin Invest 2003;111:553-9.
- Herrmann A, Sommer U, Pranada AL, Giese B, Kuster A, Haan S, et al. STAT3 is enriched in nuclear bodies. J Cell Sci 2004;117:339-49.
- Hao A, Novotny-Diermayr V, Bian W, Lin B, Lim CP, Jing N, et al. The LIM/homeodomain protein Islet1 recruits Janus tyrosine kinases and signal transducer and activator of transcription 3 and stimulates their activities. Mol Biol Cell 2005;16:1569-3.
- Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, et al. Essential role of STAT3 in cytokine-driven NF-κB-mediated serum amyloid A gene expression. Genes Cells 2005;10:1051-63.
- Pranada AL, Metz S, Herrmann A, Heinrich PC, Muller-Newen G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J Biol Chem 2004; 279: 15 114–23.
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerisation regulated by reversible acetylation of a single lysine residue. Science 2005;307:269-73.
- Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005;65:939-47.
- Sato N, Kawai T, Sugiyama K, Muromoto R, Imoto S, Sekine Y, et al. Physical and functional interactions between STAT3 and ZIP kinase. Int Immunol 2005;17:1543-52.
- Kojima H, Sasaki T, Ishitani T, Iemura S, Zhao H, Kaneko S, et al. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFβ-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc Natl Acad Sci USA 2005;102:4524-9.
- Shah M, Patel K, Mukhopadhyay S, Opavsky A, Cheng M, Welstead G, et al. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem 2006;281:7302-8.
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997;278:1803-5.
- Collum RG, Brutsaert S, Lee G, Schindler C. A Stat3-interacting protein (StIP1) regulates cytokine signal transduction. Proc Natl Acad Sci USA 2000; 97: 10 120–5.
- Zhang J, Yang J, Roy SK, Tininini S, Hu J, Bromberg JF, et al. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc Natl Acad Sci USA 2003;100:9342-7.
- Schick N, Oakeley EJ, Hynes NE, Badache A. TEL/ETV6 is a signal transducer and activator of transcription 3 (Stat3)-induced repressor of Stat3 activity. J Biol Chem 2004;279:38787-96.
- Nishimoto A, Yu Y, Lu Z, Mao X, Ren Z, Watowich SS, et al. A Ras homologue member I directly inhibits signal transducers and activators of transcription 3 translocation and activity in human breast and ovarian cancer cells. Cancer Res 2005;65:6701-10.
- Ulane CM, Kentsis A, Cruz CD, Parisien JP, Schneider KL, Horvath CM. Composition and assembly of STAT-targeting ubiquitin ligase complexes: paramyxovirus V protein carboxyl terminus is an oligomerization domain. J Virol 2005;79:10180-9.
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. β-arrestin-dependent, G-protein-independent ERK1/2 activation by the β2-adrenergic receptor. J Biol Chem 2006;281:1261-73.
- Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 1999;51:651-90.
- Chen ZJ, Minneman KP. Recent progress in α1-adrenergic receptor research. Acta Pharmacol Sin 2005;26:1281-7.
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 1998;280:574-7.
- O’Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, et al. α1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest 2006;116:1005-15.
- Zhang T, Xu Q, Chen FR, Han QD, Zhang YY. Yeast two-hybrid screening for proteins that interact with α1-adrenergic receptors. Acta Pharmacol Sin 2004;25:1471-8.
- Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, et al. Spinophilin regulates Ca2+ signaling by binding the N terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol 2005;7:405-11.
- Zhu W, Zeng X, Zheng M, Xiao RP. The enigma of β2-adrenergic receptor Gi signaling in the heart: the good, the bad, and the ugly. Circ Res 2005;97:507-9.
- Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, et al. Phospholipase Cε modulates β-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 2005;97:1305-13.
- Brixius K, Bloch W, Pott C, Napp A, Krahwinkel A, Ziskoven C, et al. Mechanisms of β3-adrenoceptor-induced eNOS activation in right atrial and left ventricular human myocardium. Br J Pharmacol 2004;143:1014-22.
- Germack R, Dickenson JM. Induction of β3-adrenergic receptor functional expression following chronic stimulation with noradrenaline in neonatal rat cardiomyocytes. J Pharmacol Exp Ther 2006;316:392-402.
- Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 2001;103:1649-55.
- Hall RA. Beta-adrenergic receptors and their interacting proteins. Semin Cell Dev Biol 2004;15:281-8.
- Gonzalez-Cabrera PJ, Gaivin RJ, Yun J, Ross SA, Papay RS, McCune DF, et al. Genetic profiling of α1-adrenergic receptor subtypes by oligonucleotide microarrays: coupling to interleukin-6 secretion but differences in STAT3 phosphorylation and gp-130. Mol Pharmacol 2003;63:1104-16.
- Yin F, Li P, Zheng M, Chen L, Xu Q, Chen K, et al. Interleukin-6 family of cytokines mediates isoproterenol-induced delayed STAT3 activation in mouse heart. J Biol Chem 2003;278:21070-75.
- Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol 2005;204:428-36.
- Yin F, Wang YY, Du JH, Li C, Lu ZZ, Han C. Noncanonical cAMP pathway and p38 MAPK mediate β2-adrenergic receptor-induced IL-6 production in neonatal mouse cardiac fibroblasts. J Mol Cell Cardiol 2006;40:384-93.
- Sasaguri T, Teruya H, Ishida A, Abumiya T, Ogata J. Linkage between α1 adrenergic receptor and the Jak/STAT signaling pathway in vascular smooth muscle cells. Biochem Biophys Res Commun 2000;268:25-30.
- Briest W, Rassler B, Deten A. Norepinephrine-induced inter-leukin-6 increase in rat hearts: differential signal transduction in myocytes and non-myocytes. Pflugers Arch-Eur J Physiol 2003;446:437-46.
- Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Semin Cell Dev Biol 2004;15:309-23.
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999;402:884-8.
- Morris JB, Pham TM, Kenney B, Sheppard KE, Woodcock EA. UTP transactivates epidermal growth factor receptors and promotes cardiomyocyte hypertrophy despite inhibiting transcription of the hypertrophic marker gene, atrial natriuretic peptide. J Biol Chem 2004;279:8740-46.
- Kim J, Eckhart AD, Eguchi S, Koch WJ. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J Biol Chem 2002;277:32116-23.
- Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of G proteins. Cell 2000;102:635-46.
- Fan G, Shumay E, Malbon CC, Wang H. c-Src tyrosine kinase binds the β2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2, and mediates agonist-induced receptor desensitization. J Biol Chem 2001;276:13240-7.
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 1999;283:655-61.
- Bence K, Ma W, Kozasa T, Huang XY. Direct stimulation of Bruton’s tyrosine kinase by G(q)-protein alpha-subunit. Nature 1997;389:296-9.
- Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem 1992;267:25798-802.
- Ho MK, Yung LY, Chan JS, Chan JH, Wong CS, Wong YH. Gα14 links a variety of Gi- and Gs-coupled receptors to the stimulation of phospholipase C. Br J Pharmacol 2001;132:1431-40.
- Lo RK, Wong YH. Signal transducer and activator of transcription 3 activation by the δ-opioid receptor via Gα14 involves multiple intermediates. Mol Pharmacol 2004;65:1427-39.
- Lo RK, Cheung H, Wong YH. Constitutively active Galpha16 stimulates STAT3 via a c-Src/JAK- and ERK-dependent mechanism. J Biol Chem 2003;278:52154-65.
- Lowry WE, Huang XY. G protein β γ subunits act on the catalytic domain to stimulate Bruton’s agammaglobulinemia tyrosine kinase. J Biol Chem 2002;277:1488-92.
- Giasson E, Servant MJ, Meloche S. Cyclic AMP-mediated inhibition of angiotensin II-induced protein synthesis is associated with suppression of tyrosine phosphorylation signaling in vascular smooth muscle cells. J Biol Chem 1997;272:26879-86.
- Sengupta TK, Schmitt EM, Ivashkiv LB. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proc Natl Acad Sci USA 1996;93:9499-504.
- Park YJ, Park ES, Kim MS, Kim TY, Lee HS, Lee S, et al. Involvement of the protein kinase C pathway in thyrotropin-induced STAT3 activation in FRTL-5 thyroid cells. Mol Cell Endocrinol 2002;194:77-84.
- Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science 2000;290:144-7.
- Pelletier S, Duhamel F, Coulombe P, Popoff MR, Meloche S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol Cell Biol 2003;23:1316-33.
- Ito M, Adachi T, Pimentel DR, Ido Y, Colucci WS. Statins inhibit beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes via a Rac1-dependent mechanism. Circulation 2004;110:412-28.
- Nakaoka Y, Nishida K, Fujio Y, Izumi M, Terai K, Oshima Y, et al. Activation of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ Res 2003;93:221-9.
- Zhang T, Ma J, Cao X. Grb2 regulates Stat3 activation negatively in epidermal growth factor signaling. Biochem J 2003;376:457-64.
- Karoor V, Wang L, Wang HY, Malbon CC. Insulin stimulates sequestration of β-adrenergic receptors and enhanced association of β-adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem 1998;273:33035-41.
- Schuringa JJ, Dekker LV, Vellenga E, Kruijer W. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase Cδ is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J Biol Chem 2001;276:27709-15.
- Wang J, Paradis P, Aries A, Komati H, Lefebvre C, Wang H. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol Cell Biol 2005;25:9829-44.
- Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, et al. Role of the protein kinase Cδ-Raf-1-EK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation 2005;112:1971-8.
- Novotny-Diermayr V, Zhang T, Gu L, Cao X. Protein kinase Cδ associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J Biol Chem 2002;277:49134-42.
- Chaturvedi P, Reddy MV, Reddy EP. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene 1998;16:1749-58.
- Huang J, Sun Y, Huang XY. Distinct roles for Src tyrosine kinase in β2-adrenergic receptor signaling to MAPK and in receptor internalization. J Biol Chem 2004;279:21637-42.
- He Q, Wu G, Lapointe MC. Isoproterenol and cAMP regulation of the human brain natriuretic peptide gene involves Src and Rac. Am J Physiol Endocrinol Metab 2000;278:E1115-23.
- Zhong H, Minneman KP. Activation of tyrosine kinases by α1A-adrenergic and growth factor receptors in transfected PC12 cells. Biochem J 1999;344:889-94.
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J 2003;374:1-20.
- Kunisada K, Hirota H, Fujio Y, Matsui H, Tani Y, Yamauchi-Takihara K, et al. Activation of JAK-STAT and MAP kinases by leukaemia inhibitory factor through gp130 in cardiac myocytes. Circulation 1996;94:2626-32.
- Ng DC, Long CS, Bogoyevitch MA. A role for the extracellular signal-regulated kinase and p38 mitogen-activated protein kinases in interleukin-1β-stimulated delayed signal transducer and activator of transcription 3 activation, atrial natriuretic factor expression, and cardiac myocyte morphology. J Biol Chem 2001;276:29490-8.
- Booz GW, Day JN, Baker KM. Angiotensin II effects on STAT3 phosphorylation in cardiomyocytes: evidence for Erk-dependent Tyr705 dephosphorylation. Basic Res Cardiol 2003;98:33-8.
- Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA 1998;95:11107-12.
- Nguyen VA, Gao B. Cross-talk between α1B-adrenergic receptor and interleukin-6 signaling pathways. Activation of α1B-AR inhibits IL-6-activated STAT3 in hepatic cells by a p42/44 mitogen-activated protein kinase-dependent mechanism. J Biol Chem 1999;274:35492-8.
- Schuringa JJ, Jonk LJ, Dokter WH, Vellenga E, Kruijer W. Interleukin-6-induced STAT3 transactivation and Ser727 phosphorylation involves Vav, Rac-1 and the kinase SEK-1/MKK-4 as signal transduction components. Biochem J 2000;347:89-96.
- Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem 2003;278:34133-40.
- Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem 2005;280:11528-34.
- Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005;129:1616-32.
- Gusterson R, Brar B, Faulkes D, Giordano A, Chrivia J, Latchman D. The transcriptional coactivators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J Biol Chem 2002;277:2517-24.