
Stearic acid protects primary cultured cortical neurons against oxidative stress1
Introduction
Epidemiological, clinical, and biochemical studies have shown that different types of dietary fatty acids can modify the risks of stroke[1,2], but the molecular and cellular mechanisms by which dietary fatty acids exert such effects are still not well understood. Free fatty acids have been focused on for their potential neuroprotection more than ever in pharmacological research. Besides being crucial biomolecules in metabolic processes, they serve as the substrates for cell membrane biogenesis (glyco and phospholipids), and the precursor molecules of intracellular signaling such as prosta-glandins, leukotrienes, thromboxanes, and the platelet activating factor. Polyunsaturated fatty acids have been implicated in the prevention of various human diseases, including obesity, diabetes, coronary artery disease, stroke, and inflammatory and neurological diseases[3]. Only a few articles have reported the effects of saturated fatty acids on human health. Saturated fats are usually regarded as unhealthy, but nutritionists believe that the type of saturated fat is also important.
Stearic acid is a long-chain fatty acid consisting of 18 carbon atoms without double bonds. It is present in fairly constant proportions in beef, pork, lamb, and veal (approximately 9%–12% of total fatty acid content), with lower proportions found in poultry (approximately 6%–7% of total fatty acid content)[4]. It is biochemically classified as a saturated fatty acid, both for the purpose of food labeling and dietary recommendations. The behavior of stearic acid is especially unique with respect to its effects on serum cholesterol levels. Studies in humans and experimental animals have suggested that ingestion of stearic acid has a neutral or cholesterol-lowering effect, which is in contrast to the effects of lauric, myristic, and palmitic acids[5]. In addition, a beneficial effect of stearic acid on clotting factors can result in a less thrombogenic state. Stearic acid, one of the most common fatty acids in brain phospholipids, originates in the circulation and is sequestered from blood by the brain along with precursor fatty acids[6]. Recently, it has been found that several fatty acids can function as natural ligands of peroxisome proliferator-activated receptors (PPAR) to regulate lipid homeostasis. PPAR are members of the nuclear receptor superfamily of ligand-dependent transcription factors. The PPAR subfamily comprises 3 isotypes: PPARα, PPARβ, and PPARγ. Although a precise biological role for PPARγ remains unclear, PPARα and PPARγ have modulatory roles in various cellular responses, including immune and inflammatory responses. Various fatty acids can regulate gene expression at micromolar concentrations through direct interactions with PPARα or PPARγ. Recently, it has been reported that ligands of PPARα or PPARγ can significantly protect the brain from ischemic or oxidative insult[7,8]. Therefore, we hypothesize that stearic acids may function as natural ligands of PPAR to protect neurons from ischemic insult. We have reported that the neuroprotective effects of stearic acid against toxicity of oxygen/glucose deprivation or glutamate on rat cortical or hippocampal slices[9]. In the present study, we evaluated the effects of stearic acid on cortical neurons insulted by glutamate NaN3 or hydrogen peroxide (H2O2).
Materials and methods
Experimental animals Pregnant Sprague-Dawley (SD) rats (300±20 g, grade II, Certification N
Reagents and drugs Poly-D-lysine, bisphenol A diglycidy ether (BADGE), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphe nyltetrazolium bromide (MTT), dizocilpine maleate (MK801), 2-thibarbituric acid (TBA), and {3-[1(p-chlorobenzyl)-5-(isopropyl)-3-t-butylthiondol-2-yl]-2, 2-dimethylpropanoic acid, Na} (MK886) were purchased from Sigma (St Louis, MO, USA). Fetal calf serum was purchased from FuMeng Gene Bio-Engi Res and Dev Co, Ltd Inc (Shanghai, China); Dulbecco’s modified Eagle’s medium (DMEM)-F12, neurobasal medium and B27 were from Gibco (Logan, UT, USA); The LDH kit, superoxide dismutases (SOD) kit, glutathione peroxidase [GSH-Px], and the catalase (CAT) kit were purchased from Jiancheng Institute of Biotechnology (Nan-jing, China). The bicinchoninic acid (BCA) protein assay kit was from Pierce (Rockford, IL, USA). Neuron-specific enolase (NSE) antibody and streptavidin biotin complex kits were purchased from Boster Biological Technology (Wuhan, China). Polyclone antibody to PPARγ and SP1 were purchased from ALEXIS Biochemicals (San Diego, CA, USA) Cytosine arabinoside was purchased from Shanghai Hua-Lian Pharmaceutical Cooperation (Shanghai, China). Stearic acid (Chemical Reagent Co, Ltd, Shanghai, China) and all other reagents were of analytical grade. Stearic acid dissolved in ethanol was diluted in culture medium before use. The final ethanol concentration in the culture medium was less than 0.5% (Table 1).

Full table
Cell culture Primary rat cultured cortical neurons were prepared from embryonic d 18 Sprague-Dawley rats. The brains were removed aseptically from the skulls, the meninges excised carefully, and the cerebral cortex was dissected in iced dissection solution, which had the following composition (in mmol/L): NaCl 113, KCl 3, CaCl2 1, MgCl2 6, NaHCO3 25, NaH2PO4 1, and glucose 11 (final pH 7.2–7.4). The bilateral cerebral cortex were collected and mechanically frag-mented, then digested with 0.25% trypsin (Sino-American Biotechnology Co, Shanghai, China), and 0.2 mg/mL DNase I (Sino-American Biotechnology Co, Shanghai, China), and incubated for 10 min at 37 ºC. Following trypsinization, the cells were washed twice in DMEM-F12 culture medium containing 15% heat-inactivated fetal calf serum. Dissociated cells were plated in 35 mm dishes or 96-well plates precoated with 0.1% poly-l-lysine at densities of 1×106/mL, 2 mL/dish, or 100 µL/well. The culture was maintained in a humidified incubator with a 5% CO2 atmosphere at 37 ºC. The medium was changed into medium consisting of neurobasal modified with B27 supplement after the cells were attached to the culture plate. After 1 d in the culture, cytosine arabinoside (5 µmol/L) was added to the medium to inhibit glial proliferation. Experiments were performed on d 6 of culturing. The purification rates were evaluated according to the cell count and NSE immunohistochemistry.
Cell viability Cell viability was evaluated by using a colorimetric assay with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphe nyltetrazolium bromide (MTT), an indicator of mitochondrial respiratory chain activity. Briefly, the cells were pretreated with stearic acid over a wide dose range at various times prior to and during subjected to glutamate, NaN3 or hydrogen peroxide insult. The viability of cells was determined using MTT assay after 24 h cotreatment. The 0.5 mg/mL MTT solution was added to each well and the cells were incubated for a further 4 h at 37 ºC. Then, the media were removed and formazan crystals were solubilized with DMSO. The optical density of each well was measured at 490 nm (OD490) by using an ELISA reader (Bio-Tek ELX800uv, Bio-Tek Instrument Inc, Winooski, VT, USA). The cells without stearic acid treatment served as the controls. The release of LDH from the intracellular compartment into the surrounding medium was expressed as an indicator of cell death. LDH activity in the extracellular medium was monitored by using a commercial kit. Data were expressed as percent of LDH release of the positive control (0.1% Triton X-100 treated group).
Glutamate uptake assay in cortical neurons[10] Cortical neurons were plated on 24-well plates. After stimulation with or without stearic acid for 24 h, the cells were washed and equilibrated in Krebs buffer solution [(mmol/L NaCl 120.6, KCl 5.9, NaH2PO4 1.2, MgCl2 1.2, NaHCO3 15.4, CaCl2 2.5, C6H12O6 11.5, pH=7.4)] for 20 min at 37 ºC. Glutamate uptake was initiated by the addition of 5 mmol/L (0.2 Ci/mmol) L-[3H] glutamate. After 10 min incubation, the reaction was stopped by washing the cells 3 times with ice-cold PBS. The cells were then lysed in 1 mol/L NaOH at 37 ºC for 30 min, and the radioactivity in the cell lysates was quantified by scintillation counting. The protein content was determined in aliquots of each lysates by the BCA method.
Lipid peroxidation[11] The lipid peroxidation of the cells was determined by measuring thiobarbituric acid reactive substances (TBARS). The cells were lysed with 4 mL fulvic acid (0.167 mol/L) and 0.5 mL 10% phosphotungstic acid, and then centrifuged at 4000×g for 10 min. The precipitation was resuspended with 1.5 mL distilled water and 0.5 mL TBA reagent (1:1, v/v) mixture of 0.67% TBA and acetic acid. The reaction mixture was heated at 95 ºC for 1 h. After cooling, 2 mL of n-butanol was added, and the mixture was shaken vigorously for 30 s. After centrifugation at 3000×g for 10 min, the n-butanol layer was used for fluorometric measurement with λex 515 nm and λem 553 nm using a fluorescence spectrophotometer. The value of fluorescence was calculated by comparing with standards prepared from 1,1,3,3-tetraethoxypropane.
Analysis of activity of antioxidant enzymes in cortical neurons For the assay of antioxidant enzymes, the cultures were washed with iced PBS buffer and then homogenized. The homogenate was centrifuged at 10 000×g for 15 min at 4 ºC. The supernatant was used as test sample for the enzyme assay. The protein concentration of the supernatant was determined by the BCA method. The total SOD and Cu/Zn SOD were measured by the guidance of the kits, respectively. The control (C) consisted of all the reagents except the supernatant (2%, w/v), while the blank (B) consisted of buffer and the supernatant without any reagents. The absorbance of T, C, and B was measured at 550 nm and the enzyme activity was expressed in units (1 U=50% inhibition of the oxidation of oxymine by the xanthine-xanthine-oxidase system). Cu/Zn SOD and MnSOD activity were distinguished by the inhibition of the former by 2 mmol/L of KCN added to the assay cocktail. Cu/Zn SOD activity was determined by subtracting MnSOD activity from the total SOD activity. Glutathione (GSH), a tripeptide antioxidant, plays multiple biological functions. It is involved in the detoxification of harmful molecules, such as reactive oxygen species (hydrogen peroxide and hydroperoxides) through glutathione peroxidase. The activity of GSH-Px was determined by quantifying the rate of H2O2-induced oxidation of reduced GSH to oxidized glutathione. A yellow product which had absorbance at 412 nm could be formed as GSH reacted with the dithiobisnitrobenzoic acid. One unit of GSH-Px was defined as the amount that reduced the level of GSH by 1 µmol/L in 1 min per mg of protein. The assay of CAT activity was based on its ability to decompose H2O2. The absorbance of the supernatant at 254 nm changed when the H2O2 solution was injected into the cuvette. The disappearance of H2O2 was measured at 240 nm for 60 s at 1 min intervals. The change of the absorbance reflected the CAT activity.
Influence of inhibitors of receptors on the neuropro-tective effects of stearic acid[9] MK886 is an indole compound that was originally identified as a potent inhibitor of the 5-lipoxygenase (5-LOX)-activating protein (FLAP). Recently, it has been found to inhibit PPARα via a non-competitive mechanism as shown by its effects on the binding of fatty acids to the PPARα protein[12]. Cortical neurons were treated with 5 µmol/L MK886 or 100 µmol/L BADGE (a PPARγ antagonist) in the presence or absence of stearic acid to determine whether antagonists of PPAR could block the neuroprotection provided by 30 µmol/L stearic acid. In addition, the influence of the protein blocker, cycloheximide (30 µmol/L), on the neuroprotective effects of stearic acid (10 µmol/L) was also investigated at the same time. Cell viability was measured by using the MTT method.
Immunoblotting of PPARγ after treatment of stearic acid Harvested cortical cells were homogenized in a buffer containing 20 mmol/L HEPES, pH 7.4, 2 mmol/L EGTA, 10 mmol/L Tris-HCl, 1 mmol/L phenylmethylsulfonyl fluoride, 2 mmol/L dithioerythreitol (DTE), and 10 µg/mL aprotinin. The cells were sonicated to disrupt the cell membranes, and the soluble (cytoplasmic) and pellet fractions were separated by centri-fugation. The homogenate was centrifuged at 100 000×g, and the soluble fraction was collected. Samples were adjusted by the addition or dilution to 0.05% Triton X-100. Equal amounts of proteins, as determined by the BCA method, were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane in a Bio-Rad (Hercules, CA, USA) transblot electrophoresis apparatus at 100 V for 2 h using Towbin's buffer (25 mmol/L Tris, pH 8.3, 192 mmol/L glycine, and 20% methanol). The membranes containing immobilized proteins were blocked with 5% non-fat milk in Tris buffer (20 mmol/L Tris, pH 7.5, and 0.5 mol/L NaCl) for 1 h. A polyclonal PPARγ that cross-reacts with both human and mouse PPAR-γ was prepared in the TS buffer and added to the transblots for overnight incubation. After washing, the membranes were incubated with a goat anti-rabbit immunoglobulin G horseradish peroxidase-conjugated antibody as the secondary antibody in the TS buffer. Immunoreactive bands were visualized by a standard enhanced chemiluminescence procedure. Sp1 levels were used as loading controls for nuclear protein expression[13].
Statistical analysis Cerebral cortexes from 10 rat fetuses were used for each experimental group, and data collected from 3–6 independent experiments were used to calculate means, which are expressed as mean±SD. SPSS statistical software 10.0 for Windows was used and statistical significance was evaluated by using one-way ANOVA and the SNK test. Statistical significance was assumed if P<0.05.
Results
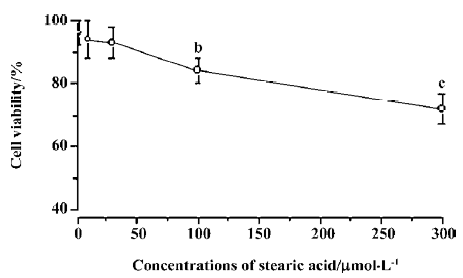
Effects of stearic acid on the viability of cortical neurons We used a NSE antibody to immunostain primary cultured cortical neurons. The healthy cortical neurons had long axons and dendrites. We could see that non-neuronal cells were very few and the purity of cultured cortical neurons was about 92%, indicating that stearic acid exhibited its effect directly, but not indirectly via non-neuronal cells (Figure 1). Stearic acid (1–30 µmol/L) did not exhibit significant cell toxicity after 24 h incubation. However, it significantly decreased cell viability after 24 h incubation when its concentrations were above 100 µmol/L. Based on our screening results, we chose concentrations of reagents that did not produce significant effects on cell viability for the experiments described below (Figure 2).
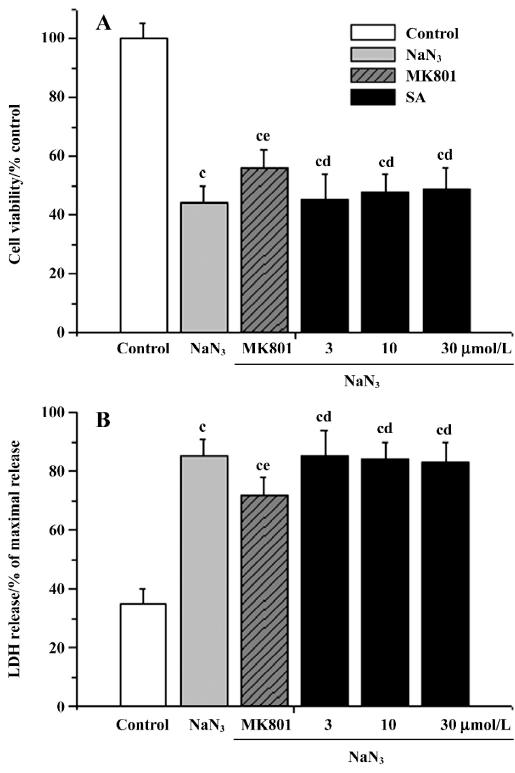
Effects of pretreatment with stearic acid on 3 different insults in cortical neurons The neuroprotective effects of stearic acid on glutamate, NaN3, or H2O2-induced neurotoxicity were investigated. The cell viability was assessed by using MTT reduction and LDH release assays. The cells were incubated with different concentrations of stearic acid for 24 h, and then exposed to 100 µmol/L glutamate, 50 µmol/L H2O2, or 20 µmol/L NaN3 for another 24 h. We found that treatment with 100 µmol/L glutamate resulted in significantly decreased cell viability (MTT 44.3%±6.4%; LDH, 78.8%±6.2%). There was a statistically significant difference between the glutamate and control groups (P<0.01). Results of both MTT reduction and LDH release assays showed that pretreatment of cortical neurons with stearic acid (3–30 µmol/L) dose-dependently prevented glutamate insult (Figure 3). After the cells were subjected to 50 µmol/L H2O2 for 24 h, cell viability was reduced by approximately 52%. H2O2 induced an increase of LDH leakage by 2.1 times than that of the control. There was a statistically significant difference between the H2O2 and control groups (P<0.01). Similarly, SA (3–30 µmol/L) protected cortical neurons against H2O2 insult in a dose-dependent manner (Figure 4). After subjected to 20 µmol/L of NaN3, cell viability was reduced by approximately 45%. NaN3 induced an increase of LDH leakage by 2.4 times than that of the control. There was a statistically significant difference between the NaN3 and control groups (P<0.01). SA (3–30 µmol/L) did not protect cortical neurons against NaN3 insult (P>0.05; Figure 5).


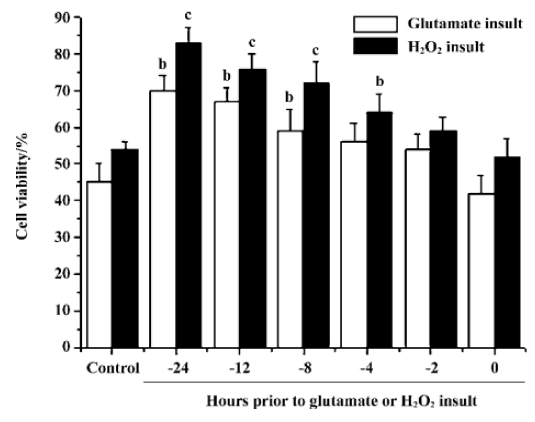
Effects of pre-incubation time on stearic acid neuroprotection of cortical neurons from glutamate or H2O2 insult We determined the minimum pre-incubation time with stearic acid needed to achieve neuroprotection. Cortical neurons required exposure to stearic acid (10 µmol/L) for 4–8 h prior to the administration of glutamate or H2O2 to achieve significant neuroprotection. Stearic acid time-dependently protected neurons against glutamate and H2O2 insults during the incubation time from 4 h to 12 h prior to the administration of insults (Figure 6).
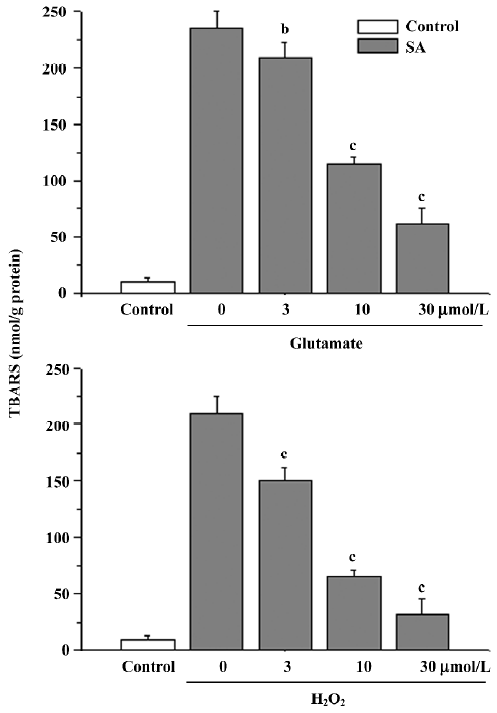
Lipid peroxidation The treatment of cells with glutamate or H2O2 caused an obvious elevation of TBARS [(200±10 nmol/g protein or 220±6 nmol/g protein), P<0.01 compared with the control group (10±4) nmol/g protein]. The elevation was inhibited by stearic acid (3–30 µmol/L; Figure 7).
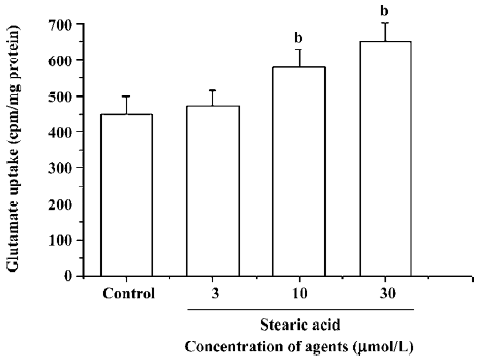
Effects of stearic acid on glutamate uptake The pretreatment of primary cortical neurons with 3–30 µmol/L of stearic acid for 24 h produced a concentration-dependent facilitation of the glutamate uptake in these cells, and reached a maximal increase of 148%±12% at 30 µmol/L. These results suggest that the effects of stearic acid on the promotion of glutamate uptake may partially contribute to its neuropro-tection (Figure 8).
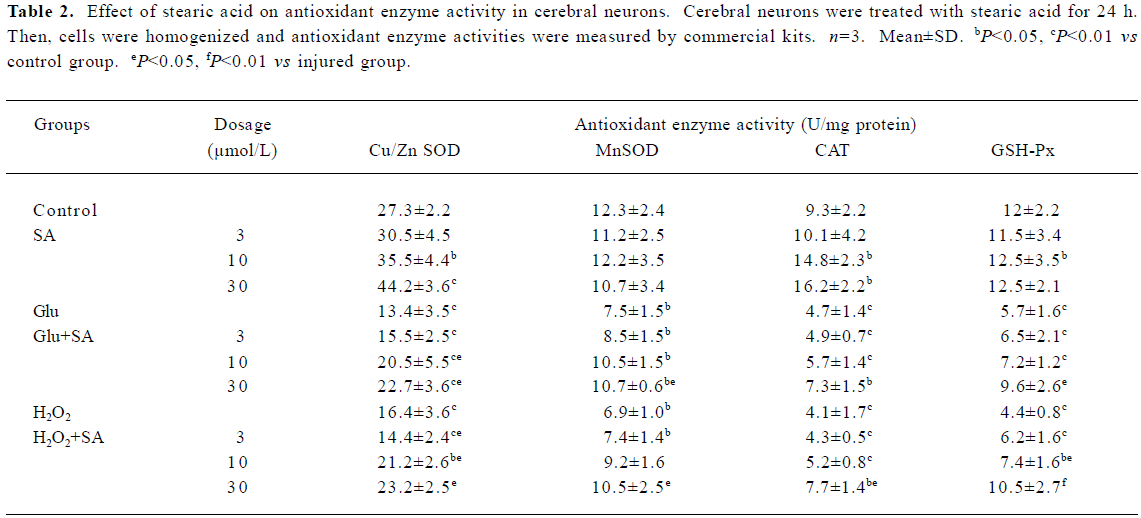
Effect of stearic acid on antioxidant enzymes Neural damage or death occurs if the increase in reactive oxygen species produced in the brain during exposure to hypoxia is not counterbalanced by an increase in the cells’ antioxidant defense systems. After incubation for 24 h, we evaluated and compared the antioxidant enzyme activity (SOD, GSH-Px, and CAT) in stearic acid groups with the corresponding control groups. Stearic acid was able to dose-dependently induce the defenses against oxidative stress through an increase in the activity of Cu/Zn SOD and CAT in cortical neurons during 24 h of incubation (Table 1). After the cells were exposed to glutamate or H2O2 insult for 24 h, obvious decreases of antioxidant enzymes, SOD, CAT, and GSH-Px were observed (P<0.01, compared with the corresponding control groups). Stearic acid dose-dependently attenuated the decrease of SOD, CAT, and GSH-Px activity (Table 2).
Full table
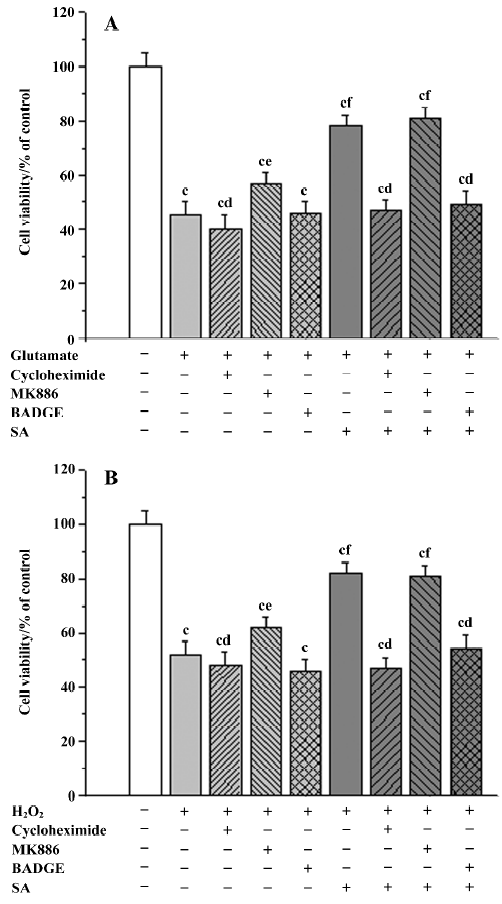
Influence of inhibitors on the neuroprotective effects of stearic acid It is known that the 5-LOX inhibitor can protect against ischemic-like injury in PC12 cells in vitro by modulating p38 MAP kinase activity[12]. Therefore, we tested whether MK886 could directly protect cortical neurons against glutamate or H2O2 insult. MK886 (5 µmol/L) at a concentration of 10 µmol/L significantly improved the activity in tissues insulted by glutamate and H2O2 (P<0.05), whereas BADGE at a concentration of 100 µmol/L produced no significant effect on cell viability insulted by glutamate or H2O2 (P>0.05). These results suggest that the inhibitor of 5-LOX directly protects tissues against oxidative stress. When neurons were treated with 5 µmol/L of MK886 during pre-incubation, the protection against glutamate or H2O2 insult afforded by stearic acid was not abolished. In contrast, 100 µmol/L BADGE blocked its protective effect completely. In addition, cycloheximide (30 µmol/L) was able to completely abolish the neuroprotective effects of stearic acid after pretreatment with cortical neurons, which suggests that the neuroprotection of stearic acid was mediated by new protein synthesis (Figure 9).
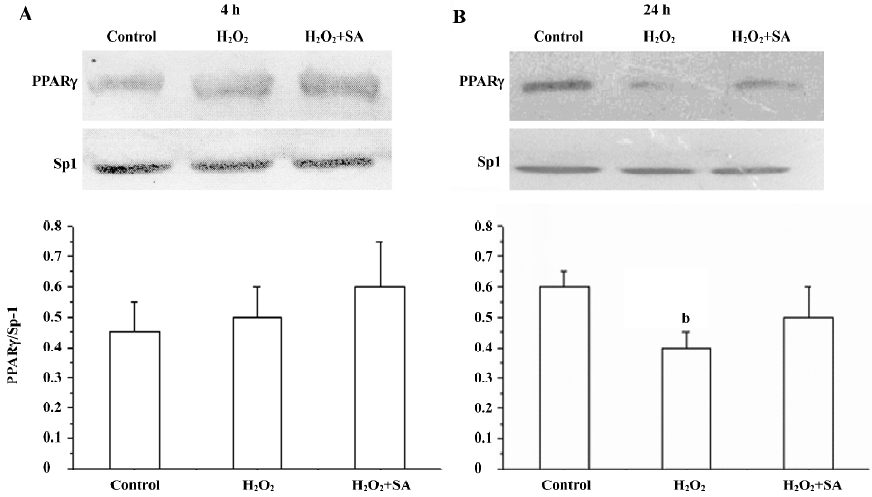
Expression of the PPARγ protein in cortical neurons It has been reported that cortical neurons expressed the PPARγ protein. Here, we investigated the effects of stearic acid on the expression of the PPARγ protein in cortical neurons during oxidative stress. Cortical neurons showed a 48 kDa band that reacted with an antibody directed at PPARγ. Our results show that the H2O2 slightly increased the expression of PPARγ after incubation for 4 h (P>0.05). However, H2O2 significantly decreased the expression of PPARγ (P<0.05) after incubation with neurons for 24 h, and the inhibitory effect of H2O2 on the expression of PPARγ can be attenuated by 30 µmol/L of stearic acid (Figure 10).
Discussion
Brain ischemia induces neuronal loss which is in part caused by excitotoxicity and free radical formation[14]. Glutamate is the primary excitatory neurotransmitter in the central nervous system. In the ischemic brain, extracellular glutamate is elevated rapidly after the onset of ischemia, which results in excessive activation of glutamate receptors and mediated neuronal injury[15]. It is known that the presence of 0.5 mmol/L glutamate in the extracellular space can reverse glutamate uptake and decrease the synthesis of glutathione in neurons[16,17].
Excitotoxic cell death is due, at least in part, to the excessive activation of ion type glutamate receptors, and hence, excessive Ca2+ influx through the receptors' associated ion channel[17]. Glutamate uptake from the extracellular space is essential to shaping excitatory postsynaptic currents and for preventing excitotoxic death due to overstimulation of glutamate receptors[15]. Stearic acid dose-dependently promoted the glutamate uptake in cerebral neurons. However, we did not find any promotion effects of stearic acid on glutamate uptake in astrocytes after incubation for 24 h (data not shown). Since stearic acid increased extracellular glutamate uptake, this property undoubtedly would help stearic acid to serve as a potent neuroprotector against glutamate toxicity in the early stages of ischemia.
Persistent activation of glutamate receptors can significantly decrease the level of GSH. Reactive oxygen species (ROS) will accumulate strikingly in a very short time as the decreasing of GSH[18]. Mitochondria are not only a source of main intracellular generators of ROS, but also the targets of ROS attack[19]. Although defenses against damage produced by ROS are extensive, including enzymatic and small molecule antioxidants as well as repair enzymes, an increased production of ROS or a poor antioxidant defense network can lead to a progressive damage in the cell with a decline in physiological function[20]. Therefore, oxidative stress-induced neuron death has been implicated in acute cerebral ischemia and chronic neurodegeneration diseases[21,22]. H2O2, a by-product of oxidative stress, has been implicated in triggering apoptosis and/or necrosis in various cell types, including cultured neurons. Exogenous hydrogen peroxide, a low molecular weight compound, can easily penetrate the lipid membrane, and cause lipid, protein, and DNA peroxidation. Here, we tested the hypothesis that stearic acid may also provide protection against oxidative stress. We found that the cortical neurons were significantly protected by stearic acid against glutamate and H2O2 insults, which suggest that neuroprotective effects of stearic acid are mediated, at least in part, by the promotion of an antioxidant mechanism. MK801, a highly potent, selective, and non-competitive NMDA receptor antagonist, can significantly protect hippocampal slices against 3 different insults (P<0.05), which suggests that persistent activation of NMDA more or less contributes to neuronal damage induced by 3 different insults.
Mitochondria are the powerhouse of the cell; their primary physiological function is to generate ATP through oxidative phosphorylation via the electron transport chain. NaN3, an inhibitor of cytochrome c oxidase and CAT, significantly decreased the viability of cerebral neurons. Although SA can dose-dependently increase the activity of CAT after 24 h incubation, SA acid did not protect cerebral neurons against NaN3 insult. These results suggest that the inhibitory effect of NaN3 on CAT activity is not counterbalanced by the promotion effects of stearic acid, and the neuropro-tection of stearic acid mainly acts on the membrane and cytoplasm instead of mitochondria during oxidative stress.
Reactive oxygen-induced free radicals play an important role as mediators of tissue injury associated with inflammatory and ischemic states. Several enzymes (SOD, GSH-Px and CAT) are important in the antioxidant defense system because they metabolize either free radicals or reactive oxygen intermediates to non-radical products. For example, SOD constitute the major enzymatic mechanism for O2–· degradation and catalyze the conversion of O2–· into H2O2. Hydrogen peroxide is in turn converted to water and molecular oxygen by CAT or GSH-Px. GSH-Px utilizes reduces GSH as the hydrogen donor. Under the same condition, the improvement of antioxidant enzymes activity closely associates with new protein synthesis or modification of their stereochemical configuration. The Cu/Zn SOD can be significantly induced by acute stressors, but not by chronic or combined stress conditions, while MnSOD can be induced under various oxidative stress and inflammatory conditions[23,24]. We precisely evaluated the effects of SA on the activity of crucial antioxidant enzymes, and found that treatment with 10 µmol/L stearic acid for 24 h significantly promoted the activity of Cu/Zn SOD in cortical neurons. In addition, we did not find any direct effective of stearic acid on GSH-Px activity after 24 h incubation. It has been reported that under oxidative stress, the activity of antioxidant enzymes increased. In our study, we found that the activity of antioxidant enzymes (SOD, CAT, and GSH-Px) in cultured cortical neurons were reduced remarkably by 24 h treatment with H2O2. The inhibitory effect of H2O2 on antioxidant enzymes could be attenuated by stearic acid, which suggests that the ability of stearic acid to preserve the antioxidant enzyme activity may contribute to its neuroprotective effect. It is known that the cytosolic form of Cu/Zn SOD appears specialized to remove superoxide produced as a result of injury. Therefore, stearic acid can only protect brain tissue viability against H2O2 insult, but NaN3 insult at a given range of concentrations and its modulatory effects on the enzyme catalytic rates are very specific and significant for each antioxidant enzyme.
In some experiments, different ligands have been used according to the different isotypes of PPAR[25–27], and these agonists all play a role in neuroprotection to different extents. Several fatty acids bind to all 3 PPAR isoforms, although there is a preference of PPARα for polyunsaturated fatty acids. We have reported that the inhibitor of PPARγ significantly blocked the neuroprotective effects of stearic acid on brain slices. Here, we determined whether inhibitors of PPAR also blocked the neuroprotection of SA on cerebral neurons. MK886 and BADGE are 2 important antagonists of PPAR. We found that BADGE completely blocked the neuropro-tective effect provided by stearic acid, whereas MK886 did not abolish its protective effect. MK886 can directly protect cortical neurons against glutamate insults. Although MK886 did not abolish the effects of stearic acid, we can not conclude that PPARα is not involved in the process of neuropro-tection. BADGE can completely block the neuroprotective effect of stearic acid, which suggests that PPARγ may play a more important role in the protective effect because BADGE has no significant effect on injured cortical neurons by itself. It has been reported that the activation of PPARγ can significantly improve Cu/Zn SOD and catalase expression in liver cells and blood vessel walls[28,29]. Our results confirm that stearic acid significantly improves antioxidant enzymes activity in cortical neurons, and its neuroprotective effects against oxidative stress have been completely blocked by the protein synthesis inhibitor after pretreatment with cycloheximide (CHX). These results strongly suggest that stearic acid significantly enhances the antioxidative enzymes activity via the activation of PPARγ and synthesis of new proteins in neural cells.
Growing evidence suggests that the expression of PPARγ in the brain is mainly involved in inflammation and neurodegeneration. Western blotting and RT-PCR results show that PPARα, PPARβ, and PPARγ are expressed in embryonic cortical neurons in vitro and that their expression is differentially modulated during their in vitro maturation. The expression of PPARα and PPARβ gradually increased during neuronal maturation, whereas the expression of PPARγ gradually decreased correspondingly during neuronal maturation[30]. A decrease in PPARγ levels might cause the development or exacerbation of inflammation[31]. Our results suggest that the ability of stearic acid to preserve the expression of PPARγ may contribute to its neuroprotective effect as well.
Stearic acid, one of the most common fatty acids in brain phospholipid, originated in the circulation and was sequestered from the blood by the brain alone with precursor fatty acids. The high concentration of stearic acid in brain gray matter suggests that this fatty acid has an important role in neural function. Very preterm babies are born with minimal fat stores and suboptimal circulating levels of these nutrients. Children with neurodevelopmental handicaps receiving adequate nourishment are generally calmer and appear more normal than those who are undernourished. Patients with less severe disabilities have an increased functional status with improved nutrition[32]. Therefore, stearic acid may effectively protect against some central nervous system injuries in preterm infants. The adult brain is typically resistant to changes in fatty acid composition; the developing brain, by contrast, is much more plastic.
Newborn animals are more tolerant than adults of cerebral hypoxia/ischemia insult, and their blood-brain barriers are easier to pass. Our results suggest that effective methods of treatment for brain ischemia may keep stearic acid at the given range of concentrations.
In conclusion, stearic acid can effectively protect cortical neurons against oxidative stress at a given range of concentrations. Its neuroprotective effect is possibly mainly mediated by the activation of PPARγ and synthesis of a new protein in cortical neurons.
References
- McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res 1999;40:1371-83.
- Needleman P, Raz A, Minkes MS, Ferrendelli JA, Sprecher H. Triene prostaglandins: prostacyclin and thromboxane biosynthesis and unique biological properties. Proc Natl Acad Sci USA 1979;76:944-8.
- Bazan NG. Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol 2005;32:89-103.
- Baer DJ, Judd JT, Kris-Etherton PM, Zhao G, Emken EA. Stearic acid absorption and its metabolizable energy value are minimally lower than those of other fatty acids in healthy men fed mixed diets. J Nutr 2003;133:4129-34.
- Monsma CC, Ney DM. Interrelationship of stearic acid content and triacylglycerol composition of lard, beef tallow and cocoa butter in rats. Lipids 1993;28:539-47.
- Tholstrup T, Marckmann P, Jespersen J, Sandstrom B. Fat high in stearic acid favorably affects blood lipids and factor VII coagulant activity in comparison with fats high in palmitic acid or high in myristic and lauric acids. Am J Clin Nutr 1994;59:371-7.
- Brune S, Kolsch H, Ptok U, Majores M, Schulz A, Schlosser R, et al. Polymorphism in the peroxisome proliferators activated receptor alpha gene influences the risk for Alzheimer’s disease. J Neural Transm 2003;110:1041-50.
- Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol 1998;161:978-84.
- Wang ZJ, Li GM, Tang WL, Yin M. Neuroprotective effects of the stearic acid against toxicity of oxygen/glucose deprivation or glutamate on rat cortical or hippocampal neurons. Acta Pharmacol Sin 2006;27:145-50.
- Miralles VJ, Martinez-Lopez I, Zaragoza R, Borras E, Garcia C, Pallardo FV, et al. Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: effect of oxidative stress. Brain Res 2001;922:21-9.
- Zhou LJ, Song W, Zhu XZ, Chen ZL, Yin ML, Cheng XF. Protective effects of bilobalide on amyloid beta-peptide 25-35-induced PC12 cell cytotoxicity. Acta Pharmacol Sin 2000;21:75-9.
- Song Y, Wei EQ, Zhang WP, Zhang L, Liu JR, Chen Z. Minocycline protects PC12 cells from ischemic-like injury and inhibits 5-lipoxygenase activation. Neuroreport 2004;15:2181-4.
- Pereira MP, Hurtado O, Cardenas A, Alonso-Escolano D, Bosca L, Vivancos J. The nonthiazolidinedione PPARgamma agonist L-796,449 is neuroprotective in experimental stroke. J Neuro-pathol Exp Neurol 2005;64:797-805.
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999;79:1431-568.
- Caccamo D, Campisi A, Curro M, Li GV, Vanella A, Ientile R. Excitotoxic and post-ischemic neurodegeneration: involvement of transglutaminases. Amino Acids 2004;27:373-9.
- Auger C, Attwell D. Fast removal of synaptic glutamate by postsynaptic transporters. Neuron 2000;28:547-58.
- Schousboe A. Transport and metabolism of glutamate and GABA in neurons and glial cells. Int Rev Neurobiol 1981;22:1-45.
- Satoh T, Ishige K, Sagara Y. Protective effects on neuronal cells of mouse afforded by ebselen against oxidative stress at multiple steps. Neurosci Lett 2004;371:1-5.
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med 2004;29:149-77.
- Ho YS, Magnenat JL, Gargano M, Cao J. The nature of antioxidant defense mechanisms: a lesson from transgenic studies. Environ Health Perspect 1998;106:1219-28.
- Sutherland BA, Rahman RM, Appleton I. Mechanisms of action of green tea catechins, with a focus on ischemia-induced neuro-degeneration. J Nutr Biochem 2006;17:291-306.
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem 2006;97:1634-58.
- Filipovic D, Radojcic MB. CuZn superoxide dismutase in the hippocampus and brain cortex of rats exposed to various stress conditions. Ann N Y Acad Sci 2005;1048:366-8.
- Lontz W, Sirsjo A, Liu W, Lindberg M, Rollman O, Torma H. Increased mRNA expression of manganese superoxide dismutase in psoriasis skin lesions and in cultured human keratinocytes exposed to IL-1 beta and TNF-alpha. Free Radic Biol Med 1995;18:349-55.
- Wayman NS, Hattori Y, McDonald MC, Filipe HM, Cuzzocreas S, Pisano B, et al. Ligands of the peroxisome proliferator-activated receptors (PPAR-γ and PPAR-α) reduce myocardial infarct size. FASEB J 2002;16:1027-40.
- Simonin MA, Bordji K, Boyault S, Bianchi A, Gouze E, Bécuwe P, et al. PPARγ-ligands modulate effects of LPS in stimulated rat synovial fibroblasts. Am J Physiol Cell Physiol 2005;282:C125-33.
- Kwak BR, Myit S, Mulhaupt F, Veillard N, Rufer N, Roosnek E, et al. PPARγ but not PPARα ligands are potent repressors of major histocompatibility complex class II induction in atheroma-associated cells. Circ Res 2002;90:356-62.
- Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D, et al. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke 2005;36:353-9.
- Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol 2002;16:2793-801.
- Cimini A, Benedetti E, Cristiano L, Sebastiani P, D'Amico MA, D'Angelo B, et al. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRs) in rat cortical neurons. Neuroscience 2005;130:325-37.
- Raikwar HP, Muthian G, Rajasingh J, Johnson C, Bright JJ. PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J Neuro-immunol 2005;167:99-107.
- Uauy R, Castillo C. Lipid requirements of infants: implications for nutrient composition of fortified complementary foods. J Nutr 2003;133:2962S-72S.