
Formation of 4'-carboxyl acid metabolite of imrecoxib by rat liver microsomes1
Introduction
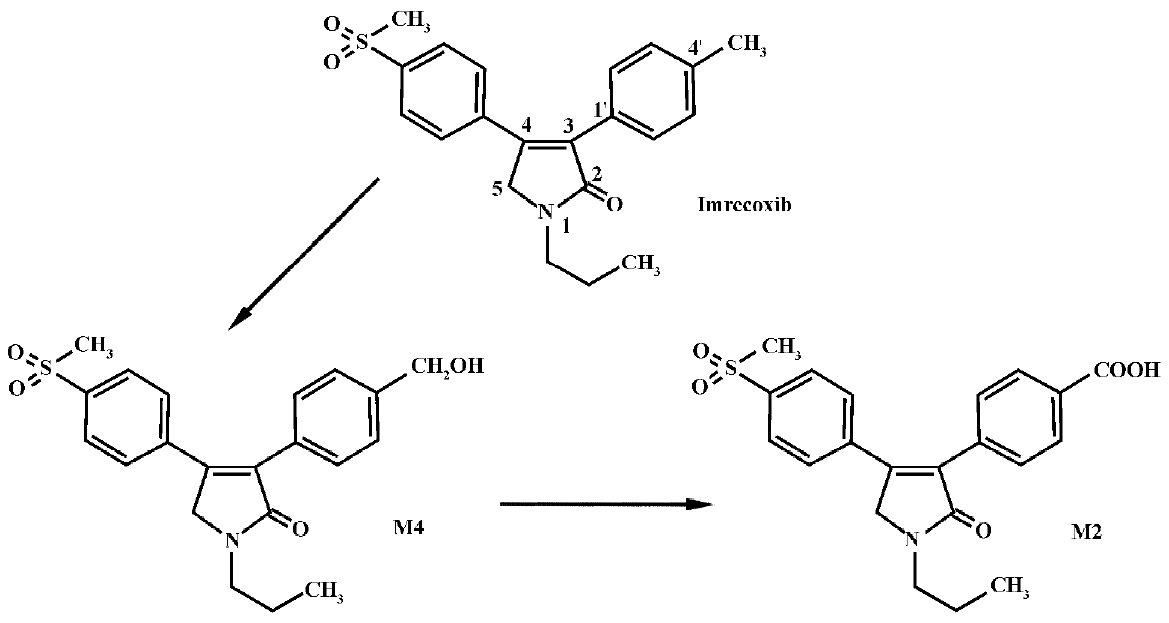
Imrecoxib [4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydro-pyrrol-2-one] (Figure 1) is a novel and moderately selective inhibitor of COX-2[1−3], an inducible isoform of cyclooxygenase that plays a key role in inflammatory processes. The drug inhibits COX-1(determination for 6-keto-PGF1a) and COX-2 (determination for PGE2) with IC50 values of 115±28 nmol/L and 18±4 nmol/L, respectively. It produces an anti-inflammatory effect by inhibiting COX-2 mRNA expression[3]. Imrecoxib has been introduced into clinical trials in China for the treatment of acute and chronic inflammatory disease. In a previous study (unpublished data), we found that imrecoxib was extensively metabolized after iv administration, with less than 2% of the dose recovered in urine or feces as parent drug. The major route of metabolism appears to be methyl hydroxylation, followed by further oxidation to 4'-carboxylic acid imrecoxib (Figure 1). Approximately 75% of the dose was excreted as the 4'-carboxylic acid metabolite of imrecoxib (M2) and approximately 15% of the dose was excreted as the 4'-hydroxymethyl metabolite (M4). M2 was mainly excreted in feces, whereas M4 was primarily excreted in urine. In vitro investigation showed that M4 was the major metabolite detected in rat liver microsomal incubates when 200 mmol/L of imrecoxib was used as a substrate, but no M2 was observed[4]. Moreover, cytochrome P450 (CYP) 3A and 2D appear to be the major P450 enzymes involved in the initial methyl hydroxylation of imrecoxib in rat. The primary objective of the present study was to investigate the formation of M2 and to characterize the enzyme(s) involved in the metabolic pathway in rats.
Materials and methods
Chemicals Imrecoxib, 4'-hydroxymethyl (M4) and 4'-carboxylic acid (M2) metabolites of imrecoxib, and BAP 910 (an analogue of imrecoxib, used as internal standard) were generous gifts from the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). α-Naphthoflavone, β-naphtho-flavone, methylpyrazole, and quinine were all purchased from Sigma-Aldrich (Deisenhofen, German). Cimetidine was provided by Kaili Pharmaceutical Co (Jiangsu, China), ketocona-zole by Dragon Pharmachemical Co (Zhejiang, China), dexamethasone by Tianjin Pharmaceutical Group (Tianjin, China), isoniazid by Jiangbei Pharmaceutical Co (Zhejiang, China), and phenobarbital sodium by Xinya Pharmaceutical Co (Shanghai, China). β-Nicotinamine adenine dinucleotide phosphate (reduced form, NADPH) was obtained from Xinjingke Biotechnology (Beijing, China). Nicotinamine adenine dinucleotide (reduced form, NADH), DL-dithio-threitol (DTT, ultra-pure grade) and tris (hydroxy-methyl) aminomethane (Tris, ultra-pure grade) were purchased from Ameresco, USA. Methanol and acetonitrile were of high performance liquid chromatography grade (Yuwang, Shan-dong, China). All other chemicals were of analytical grade.
Treatment of animals Thirty male Wistar rats (7–8 weeks old) weighing 200–250 g were supplied by the Laboratory Animal Center of Shenyang Pharmaceutical University (grade II, certificate N
Preparation of rat liver microsomes Pooled liver microsomes from 6 rat livers in each group were prepared as previously described[5]. The cytosolic protein fraction was isolated as the supernatant resulting from the first ultracentrifugation and stored at -80 °C until used. The microsomal and cytosolic protein concentrations were determined by using the method of Lowry et al[6]. Total cytochrome P450 content was measured according to the method of Omura and Sato[7].
In vitro incubation procedures To investigate the formation of 4'-carboxylic acid imrecoxib (M2), 4'-hydroxy-methyl imrecoxib (M4) (20−800 µmol/L) or 600 µmol/L imrecoxib was incubated with cytosol (1 g/L, 3 g/L, or 10 g/L) in Tris-HCl buffer (pH 7.4), which contained 1.0 mmol/L NADPH, 10 mmol/L KCl and 10 mmol/L MgCl2, in the presence or absence of 1.0 g/L microsomes, or incubated with 1.0 g/L microsomes only. The final volume was 400 μL and the mixtures were incubated at 37 °C for various times (0, 20, 40, 60, or 120 min). The reactions were initiated by addition of NADPH after a 5 min preincubation at 37 °C, and were terminated with 100 μL of cooled methanol. Then 40 μL internal standard (BAP 910, 10 μmol/L in methanol) and 400 μL of 20 mmol/L NH4H2PO4 buffer (pH 3) were added to the mixtures. The samples were extracted with 2 mL of ethyl acetate by shaking for 10 min at room temperature, and the supernatant was evaporated to dryness under a stream of nitrogen at 40 °C. The residue was dissolved in 100 μL of the mobile phase for LC/MSn assay. The dependence of M2 formation on the cofactor NADH (1.0 mmol/L) was also evaluated. Controls were conducted in the same manner, except for the absence of the NADPH or NADH. Blank samples were assayed without substrate.
In order to characterize CYP isozymes involved in the formation of M2 from M4, untreated and dexamethasone-induced microsomal samples were pretreated with selective P450 inhibitors, α-naphthoflavone (CYP 1A), quinine (CYP 2D), methylpyrazole (CYP 2E), cimetidine (CYP 2C), and ketoconazole (CYP 3A), at three inhibitor concentrations in the range of 2–50 µmol/L. The substrate concentration of M4 was 200 µmol/L. Incubations were performed at 37 °C for 40 min. As all the inhibitors were dissolved in methanol, an equivalent volume of methanol (without inhibitors) was included in the control incubations to correct for any effects of the solvent on microsomal activity. The concentration of methanol was 0.5%. The reactions were initiated by addition of NADPH after a 5 min preincubation, and were terminated with 100 µL of cooled methanol. The remaining part of the procedure was performed as described earlier for the samples without inhibitors.
LC/MSn analysis The formed M2 was quantified by a slight modification of the LC/MSn method described previously[4]. A mobile phase consisting of methanol–20 mmol/L ammonium acetate–formic acid (70:30:0.5; v/v/v) was used. Quantification was performed using selected reaction monitoring (SRM) of the transitions of m/z 400 → m/z 358, 278 for M2 and m/z 374 → m/z 278 for BAP 910 (IS), respectively. All data were analyzed by using Xcalibur software (version 1.2, Thermo Finnigan MAT). A calibration curve in the concentration range of 0.05–10.0 μmol/L for M2 was established. The precision was expressed by relative standard deviation (RSD) and the accuracy by relative error (RE%). The intra-day and inter-day precision were all less than 9.6%. The accuracy ranged from -10.1% to 6.3%.
Data analysis All data represent the means obtained from 3 individual incubations. Significant difference of a value compared with the control was determined using ANOVA followed by a one-tailed Student’s t-test.
Apparent Km and Vm values for CYP-mediated metabolism were obtained by applying a nonlinear least-squares program, MULTI[8]. Data were also analyzed by linear transformation (Eadie-Hofstee plot) to confirm a single Km model.
Results
Studies with subcellular fractions Our previous study showed that no M2 was formed in rat liver microsomes when 200 µmol/L imrecoxib was used as substrate[4]. We speculated that the generation of M2 in vitro may require the presence of cytosolic enzymes as in the metabolism of celecoxib[9]. Sandberg et al reported that alcohol dehydrogenase in cyto-sol was involved in the biotransformation of hydroxyl-celecoxib to carboxylcelecoxib[9]. So the formation of M2 was initially studied using M4 as substrate. M4 (400 µmol/L) was first incubated with liver cytosol (1 g/L, 3 g/L, or 10 g/L) from untreated rats and induced rats at 37 °C for 60 min. The reactions were initiated by addition of NADPH or NADH alone, or both NADPH and NADH as cofactors (Figure 2). Because almost no M2 formation was detected in all of these samples, corresponding liver microsomes were added to the incubation medium. This caused a marked increase in the rate of M2 formation in untreated and induced rat samples, when NADPH alone or both NADPH and NADH were used as cofactors. The formation rates of M2 in the samples using both NADPH and NADH as cofactors were similar to those in the corresponding samples using NADPH alone as cofactor. This suggests that NADH did not change the biotransformation extent of M2 when it was combined with NADPH as incubation cofactors. Surprisingly, almost no M2 was measured in the incubations when NADH was used as the sole cofactor. With all the results mentioned here, it was evident that the oxidation of M4 to M2 only required microsomes together with NADPH as cofactor.
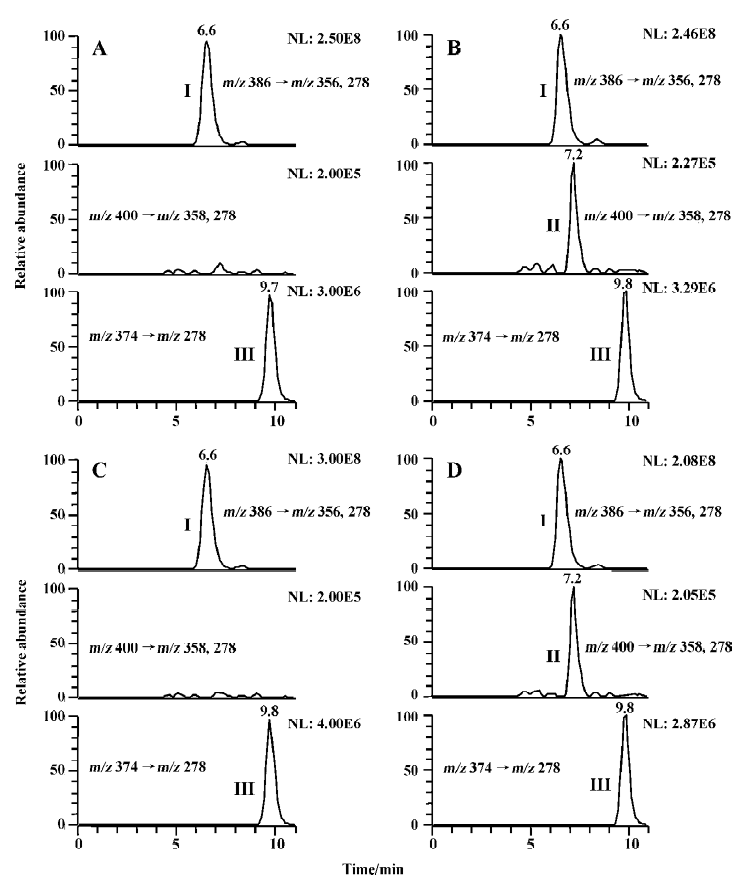
Incubation of M4 with microsomes alone was also studied. M4 400 µmol/L was incubated at 37 °C for 40 min with induced and untreated rat liver microsomes in the presence of NADPH. The formation rates of M2 in untreated, β-naphthoflavone-induced, isoniazid-induced, phenobarbital-induced, and dexamethasone-induced rat liver microsomes were 6.0±1.2, 6.3±0.9, 5.3± 0.6, 25.1±2.3, and 173.2± 37.7 pmol/min per mg protein, respectively. β-Naphthoflavone is a well-known inducer of CYP 1A, whereas isoniazid and dexamethasone are often used for the induction of CYP 2E and 3A, respectively[10,11]. Phenobarbital has been reported to be a typical inducer for CYP 2B, whereas CYP 3A also has elevated levels in phenobarbital-induced animals[12,13]. Among these typical P450 inducers, dexamethasone caused the most induction of M2 formation, followed by phenobarbital, whereas β-naphthoflavone and isoniazid had no significant effect on the reaction. Although the results with phenobarbital induction did not completely exclude the possibility of CYP 2B contributing to the reaction, the results clearly showed that CYP 3A had an important role for the formation of M2 in rat liver microsomes, whereas the contribution of CYP 1A, and 2E may not have been substantial in the reaction. Therefore, the following inhibition study and enzyme kinetic study was only carried out using untreated and dexamethasone-induced rat liver microsomes.
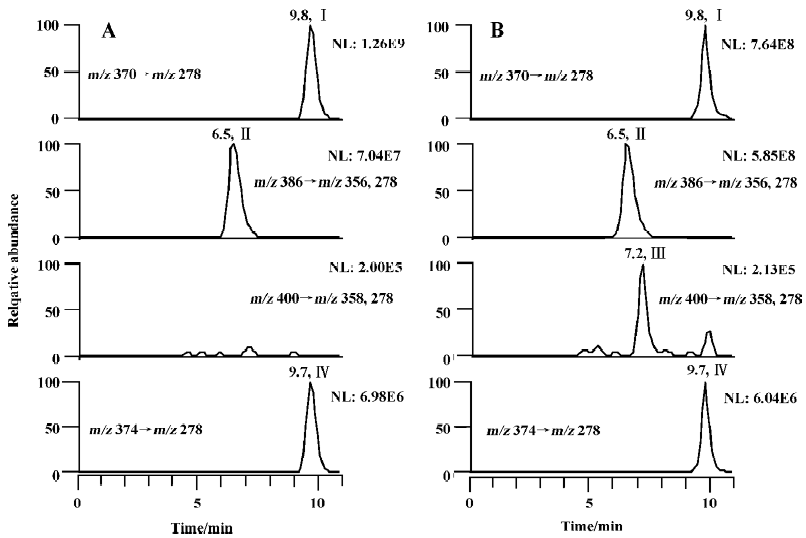
The metabolism of imrecoxib in cytosol was also investigated in the present study. When 600 µmol/L of imrecoxib was incubated with cytosol alone at 37°C for 120 min (the final volume of incubation was 1 mL), no M4 and M2 were detected in all kinds of cytosol samples, whether NADPH or NADH alone, or both NADPH and NADH was used as cofactors. Because the result of the biotransformation of M4 to M2 showed that the formation of M2 occurred in microsomes, whereas previous study showed that no M2 was formed in rat liver microsomes when imrecoxib was used as substrate[4], incubation condition was modified in the present study to investigate the formation of M2 from imrecoxib in rat liver microsomes. Microsomal incubations were carried out using 600 µmol/L of imrecoxib as substrate incubated at 37 °C for 120 min. Under these conditions, no M2 but only M4 was detected in untreated, β-naphtho-flavone-induced, isoniazid-induced, and phenobarbital-induced microsomal incubates (Figure 3), but as shown in Figure 3B, a small peak for M2 was detected in dexamethasone-treated rat liver microsomal samples. Then the effects of liver cytosol and cofactor NADH on the formation of M2 from imrecoxib were studied by adding corresponding cytosol (1 g/L, 3 g/L, or 10 g/L) and NADH (1.0 mmol/L) to the microsomal incubation medium. However, in untreated rat samples containing both microsomes and cytosol, still no M2 formation was observed, although NADPH and NADH were both used as cofactors. The same result was obtained in b-naphthoflavone-induced, isoniazid-induced, and phenobarbital-induced rat incubations that contained both microsomes and cytosol. In dexamethasone-induced rat samples, the rate of M2 formation was not increased by addition of cytosol and NADH to the microsomal incubation medium.
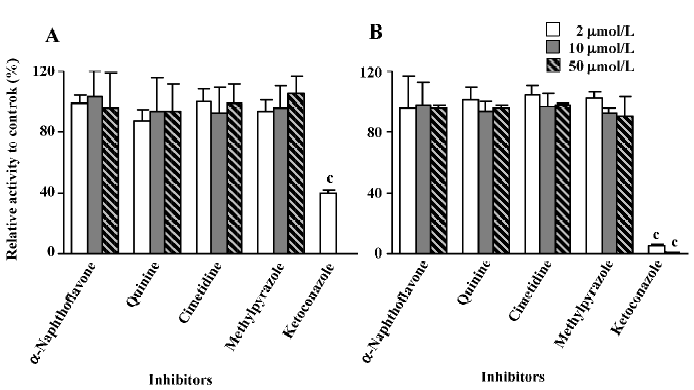
Inhibition studies The inhibitory activities were expressed as a ratio in comparison with the control activity without inhibitors. Ketoconazole, a selective inhibitor of CYP 3A, was shown to effectively decrease the formation rate of M2. When 2 μmol/L of ketoconazole was used, the reaction activity was reduced to approximately 40% of the control activity in microsomes from untreated rats, whereas only 5.6% of the control activity remained in microsomes from dexamethasone-treated rats at the same ketoconazole concentration. When the concentration of ketoconazole was increased to 10 μmol/L, no M2 was formed in untreated rat liver microsomes (the determined concentration of M2 was lower than the LLOQ of the analysis method, so the formed amount of M2 was recorded as zero) and less than 1% of the control activity remained in dexamethasone-induced rat liver microsomes. When 50 μmol/L of ketoconazole was used, M2 could not be detected in either the untreated rat liver microsomes or the dexamethasone-treated rat liver micro-somes. In contrast, other chemical inhibitors, α-naphtho-flavone, cimetidine, quinine, and methylpyrazole, did not produce any significant effects on the reaction (Figure 4).
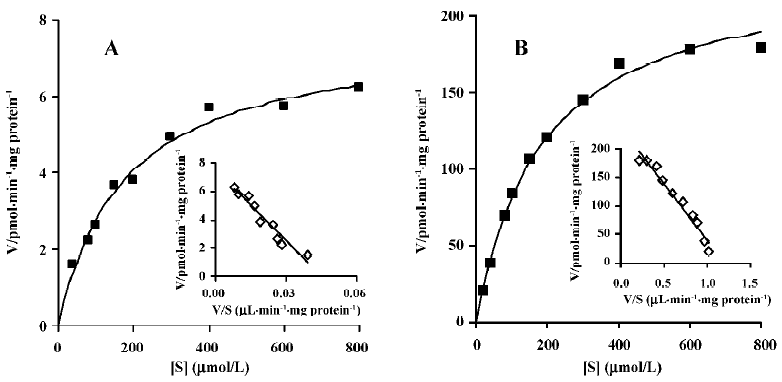
Enzyme kinetic study Linear conditions for the formation of M2 from M4 were established with respect to microsomal protein content and incubation time. The rate of formation was linear over 60 min incubation and 0.5–1.5 g/L of microsomal protein. The Michaelis-Menten kinetics of the biotransformation of M4 to M2 by rat liver microsomes was determined using 10 substrate concentrations in the range of 20–800 μmol/L at 37 °C for 40 min. The concentration of M2 formed by untreated rat liver microsomes was too low to be detected when the substrate concentration was 20 mmol/L, so apparent Km and Vm in untreated rat liver microsomes were calculated using 9 substrate concentrations. Eadie-Hofstee plots for the formation of M2 from M4 shown in Figure 5 exhibited monophasic behavior, suggesting that a single isoform of CYP may be involved in the oxidation of M4 to M2 in rat liver microsomes. Accordingly, a simple Michaelis-Menten kinetic analysis was used to estimate Km and Vm. The values of Km and Vm in untreated rat liver microsomes were 178.2 μmol/L and 7.7 pmol/min per mg protein, respectively. The corresponding values in dexamethasone-treated rat liver microsomes were 187.0 μmol/L and 233.6 pmol/min per mg protein, respectively (Table 1).

Full table
Discussion
The formation of M2 and the CYP isozymes involved were investigated in the present study. No M2 was formed in untreated, β-naphthoflavone-induced, isoniazid-induced, or phenobarbital-induced rat liver microsomes when imrecoxib was used as substrate. However, the formation of M2 was obvious in rat liver microsomal incubates with M4 used instead of imrecoxib as substrate. Moreover, dexamethasone considerably increased the formation rates of M4 from imrecoxib[4] and of M2 from M4, so a small amount of M2 was detected in dexamethasone-induced rat liver microsomes when higher concentration of imrecoxib (600 μmol/L) and longer incubation time (120 min) were used. That no formation of M2 was detected in untreated, β-naphthoflavone-induced, isoniazid-induced, and phenobarbital-induced rat liver microsomes when imrecoxib was used as substrate may be due to the small amount of M4 formed from imrecoxib and the low formation rate of M2 from M4 in these microsomes.
To identify the CYP isozymes involved in the formation of M2 from M4, the effects of specific inhibitors of CYP on this reaction were examined. The biotransformation of M4 to M2 was inhibited strongly and concentration-dependently by ketoconazole (Figure 4), a well-known inhibitor of CYP 3A[11,14]. However, in the enzyme kinetic study, Vm of M2 in dexamethasone-treated microsomes was 30-fold higher than that in untreated microsomes (Table 1). This shows that the formation rate of M2 was considerably increased by dexamethasone-induced rat liver microsomes, compared with untreated microsomes, and that dexamethasone is a specific inducer for CYP 3A[10]. These results indicate that CYP 3A is the principal enzyme involved in the biotransformation of M4 to M2.
Quinine (a inhibitor specific for CYP 2D[11,15]) and cimetidine (often used as an inhibitor for CYP 2C[15,16]) did not decrease the formation rate of M2 significantly, so the involvement of CYP 2D and 2C in the formation of M2 from M4 seems unlikely. α-Naphthoflavone and methylpyrazole were used to confirm the participation of CYP 1A[11,14,15] and CYP 2E[14] in the reaction, respectively; however, the formation of M2 was not inhibited by cimetidine and methylpyrazole in microsomes either from untreated rats or from dexamethasone-treated rats. Moreover, the formation rates of M4 in untreated, β-naphthoflavone-induced, and isoniazid-induced rat liver microsomes were 6.0±1.2, 6.3±0.9, and 5.3± 0.6 pmol/min per mg protein, respectively. These results indicate that the inducer of CYP 1A, β-naphthoflavone, and the inducer of CYP 2E, isoniazid, did not significantly increase the formation rate of M2. Therefore, substantial participation of CYP 1A and 2E may be excluded.
In conclusion, the biotransformation study of imrecoxib in rat liver subcellular fractions indicates that the second-step oxidation of imrecoxib occurs in rat liver microsomes and is NADPH-dependent. The reaction was mainly catalyzed by CYP 3A in untreated rats and in dexamethasone-induced rats. Other CYP, such as CYP 1A, 2C, 2D, and 2E, seem unlikely to participate in the metabolic pathway from M4 to M2.
Acknowledgements
We would like to thank Dr Xue-qing LI (Drug Metabolism and Pharmacokinetics and Bioanalytical Chemistry, AstraZeneca Research and Development, Mölndal, Sweden) and Prof Shu-qiu ZHANG (Department of Pharmaceutics, Shanxi Medical University, Shanxi, China) for their advice and assistance.
References
- Bai AP, Guo ZR, Hu WH, Shen F, Cheng GF. Design, synthesis and in vitro evaluation of a new class of novel cyclooxygenase-2 inhibitors: 3,4-diaryl-3-pyrrolin-2-ones. Chin Chem Lett 2001;12:775-8.
- Shen F, Bai AP, Guo ZR, Cheng GF. Inhibitory effect of 3,4-diaryl-3- pyrrolin-2-one derivatives on cyclooxygenase 1 and 2 in murine peritoneal macrophages. Acta Pharmacol Sin 2002;23:762-8.
- Chen XH, Bai JY, Shen F, Bai AP, Guo ZR, Cheng GF. Imrecoxib: a novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004;25:927-31.
- Xu HY, Xie ZY, Zhang P, Sun J, Chu FM, Guo ZR, et al. Role of rat liver cytochrome P450 3A and 2D in the metabolism of imrecoxib. Acta Pharmacol Sin 2005;27:372-80.
- Vonbarhr C, Groth CG, Jansson H, Lundgren G, Lind M, Glaumann H. Drug metabolism in human liver in vitro: establishment of a human liver bank. Clin Pharmacol Ther 1980;27:711-25.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-75.
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 1964;239:2370-8.
- Yanaoka K, Tanigawara Y, Nakagawa T, Iga T. A pharmacokinetic analysis program (MULTI) for microcomputer. J Pharmacobio-Dyn 1981;4:879-85.
- Sandberg M, Yasar U, Stromberg P, Hoog JO, Eliasson E. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol degydrogenase. Br J Clin Pharmacol 2002;54:423-9.
- Yamazaki H. Rat cytochrome P450 1A and 3A enzymes involved in bioactivation of tegafui to 5-fluorouracil and autoinduced by tegafur in liver microsomes. Drug Metab Dispos 2001;29:794-7.
- Szotakova B, Skalova L, Baliharova V, Dvorscakova M, Storkanova L, Sispera L, et al. Characterization of enzymes responsible for biotransformation of the new antileukotrienic drug quinlukast in rat liver microsomes and in primary cultures of rat hepatocytes. J Pharm Pharmacol 2004;56:205-12.
- Heuman DM, Gallagher EJ, Barwick JL, Wlshourbagy NA, Guzelian PS. Immunochemical evidence for induction of a common form of hepatic cytochrome P450 in rats treated with pregnenolone-16-carbonitrile or other steroidal or non-steroidal agents. Mol Pharmacol 1982;21:753-60.
- Kocarek TA, Schuetz EG, Strom SC, Fisher RA, Guzelian PS. Comparative analysis of cytochrome P4503A induction in primary cultures of rat, rabbit, and human hepatocytes. Drug Metab Dispos 1995;23:415-21.
- Paul LD, Springer D, Staack RF, Kraemer T, Maurer HH. Cytoxhrome P450 isoenzymes involved in rat liver microsomal metabolism of californine and protopine. Eur J Pharmcol 2004;485:69-79.
- Shimizu M, Matsushita R, Matsumoto Y, Fukuoka M. 4'-Hydroxylation of flurbiprofen by rat liver microsomes in fasting and feeding conditions. Biol Pharm Bull 2003;26:1448-54.
- Szakacs T, Veres Z, Vereczkey L. Effect of phenobarbital and spironolactone treatment on the oxidative metabolism of antipyrine by rat liver microsomes. Pol J Pharmacol 2001;53:11-9.