
Interaction between Cl– channels and CRAC-related Ca2+ signaling during T lymphocyte activation and proliferation1
Introduction
Activation of T lymphocytes with mitogen results in a biphasic elevation of intracellular calcium ([Ca2+]i) due to Ca2+ release from internal stores followed by a sustained Ca2+ influx across the plasma membrane[1,2]. The latter phase is critical for subsequent calcineurin activation and interleukin (IL)-2 gene expression in activated T cells[3,4]. The voltage-independent calcium release activated Ca2+ (CRAC) channels have been strongly suggested to be solely responsible for sustained Ca2+ influx during mitogenic stimulation of T cells by lectins or antibodies[2–4]. The evidence indicating that concanavalin (ConA) induces Ca2+ influx through CRAC channels includes: (1) the Ca2+ currents induced by mitogen stimulation and by passive store depletion [using intracellular ethyleneglycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA) or an inhibitor of endoplasmic reticulum Ca2+-ATPases that deplete internal Ca2+ stores and irreversibly activate store-operated Ca2+ (CRAC) channels in the plasma membrane] share the same properties, including ion selectivity, conductance, Ca2+-dependent inactivation, and blockage by heavy metals (Ni2+, Co2+, Mn2+, Cd2+)[3,5]; (2) stimulation of T cells with lectins or antibodies does not further increase Ca2+ influx or Ca2+ current in T cells whose stores are already depleted by thapsigargin (TG)[3,6]; (3) 1-{beta-[3-(4-methoxyphenyl) propoxyl]-4-methoxy-phenethyl}-1H-imidazole (SK&F96365), a CRAC channel blocker, blocks ICRAC in parallel with the [Ca2+]i rise and induction of T cell activation markers by TG[4]; and (4) a specific defect in Ca2+ influx associated with the absence of ICRAC was shown in nonresponsive T cells from patients with a severe immunodeficiency[7], and, likewise, T cell activation with anti-CD3 evokes Ca2+ release but not influx in Jurkat mutants selected specifically for the absence of store-operated Ca2+ entry[8].
Cl– channels are involved in the proliferation of a variety of cell types, such as endothelial cells, glioma cells, hepato-cytes, and vascular smooth muscle cells[9–14]. Previous studies (including our own) have also shown that T cell activation and proliferation were inhibited by Cl– channel blockers [4,4'-diisothiocyanostilbene-2,2'-disulfonic acid (DIDS), 5-nitro-2-(5-phenylpropylamino)-benzoate (NPPB) and tamoxifen] as well as by antisense oligonucleotides against ClC-3[15–19]. We hypothesized that these Cl– channel blockers affected T cell proliferation through Ca2+-signaling and the interaction between Cl– channels and CRAC channels that was involved in T lymphocyte activation and proliferation.
The present work was designed to further examine several key events in ConA-induced T cell activation and proliferation, including Ca2+ signaling, IL-2 mRNA expression, and final T cell proliferation. The functional role of Cl– and CRAC channels in T cell activation was confirmed by a [3H]thymidine proliferation assay using several kinds of channel blockers. To investigate whether there is an interaction between Cl– and CRAC channels in stimulated T cells, we further compared the effects of a combination of these channel blockers and each alone on ConA-induced Ca2+ signaling, IL-2 mRNA expression, and final T cell proliferation by using [3H]thymidine incorporation, Fura-2 analysis of Ca2+ movement, RNase protection assay (RPA) of human IL-2 mRNA expression and reverse transcription-polymerase chain reaction (RT-PCR) analysis of ClC-3 mRNA expression. Our data suggest an interplay between DIDS-sensitive Cl– channels and CRAC-related Ca2+ signaling during T cell activation and proliferation.
Materials and methods
Isolation of human T lymphocytes Human venous blood was obtained from the Guangzhou Blood Center and T lymphocytes were isolated as described previously[20]. Briefly, venous blood from healthy adult donors was collected in heparinized tubes and diluted with RPMI-1640 containing 25 mmol/L N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), 10% heated-activated fetal calf serum (FCS), 2 mmol/L glutamine, 50 mg/mL streptomycin, and 50 U/mL penicillin. Peripheral blood mononuclear cells were centrifuged on Ficoll-Paque (Shanghai Reagent Institute, Shanghai, China), and then transferred to a sterile nylon wool fiber column (Polysciences Inc, Warrington, USA) pre-equilibrated with RPMI-1640/10% FCS at 37 °C for 45 min. The column was eluted with pre-warmed RPMI-1640/10% FCS, and the cells were washed 3 times with this medium. Cell populations obtained in this way were ≥95% T lymphocytes, which were measured by fluorescence-activated cell sorter analysis with anti-CD3 antibody OKT3 (Ortho Pharmaceuticals, Raritan, NY, USA).
Proliferation assay Fresh isolated T lymphocytes were suspended in RPMI-1640/FCS and 2×105 cells were placed in each well of a 96-well plate. The cells were preincubated with the compounds tested for 15 min and then exposed to mitogenic lectin and ConA (5 µg/mL). At a concentration of 5 µg/mL, ConA induces significant proliferation of isolated human T lymphocytes without any toxic effects, which has been confirmed in many previous studies[1–3,15,16,18,21–23]. The cultures were maintained in a humidified incubator at 37 °C (in 5% CO2) for 72 h. Sixteen hours before the cells were harvested, 18.5×103 Bq [3H]thymidine (Shanghai Atomic Energy Research Institute, Shanghai, China) was added to each well. The incorporated radioactivity was measured in a Beckman scintillation counter (Beckman, USA), expressed as counts per minute (3 replicates/experiment). In order to exclude the direct cytotoxic effects of the tested compounds on T cells, the trypan blue exclusion assay was used to determine cell viability at the end of incubation, and only samples with a viability of at least 95% were used for further assay. At the concentration used, no cytotoxic effect on cell cultures was found.
[Ca2+]i measurements [Ca2+]i was measured in a T lymphocyte suspension with a Fura-2 fluorescent probe using a well-established method as described previously[13]. Briefly, fresh isolated T lymphocytes (1×108 cells/L) were incubated in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium containing Fura-2/AM (1 µmol/L) for 30 min at room temperature. T cells were diluted with RPMI-1640/10% FCS (1:3) and kept at room temperature for 10 min to allow re-equilibration. Cells were washed twice with a Ca2+-balanced salt solution (BSS; in mmol/L: NaCl 130, KCl 5, MgCl2 1, CaCl2 1.5, HEPES 20, glucose 10, pH 7.4), and finally suspended in BSS (2×106−4×106 cells/mL). [Ca2+]i was monitored by using a fluorescence spectrophotometer (RF-5000; Shimadzu, Kyoto, Japan) with dual excitation at 340 nm/380 nm and emission at 500 nm. In order to chelate or remove the residual Ca2+ in Ca2+-free medium, EGTA (50 mmol/L) was added into Ca2+-free medium 1 min before determining the release of intracellular Ca2+. [Ca2+]i was determined from the relation: [Ca2+]i =Kd×Sf380/b380 (R–Rmin)/(Rmax–R), where Kd is 224 nm in the cytoplasmic environment; Sf380/b380 is the ratio of the intensities of the free and bound dye forms at 380 nm; R is the fluorescence ratio (340 nm /380 nm) of the intracellular Fura-2; Rmax and Rmin are the maximal and minimal fluorescence ratios obtained by addition of Triton X-100 (final concentration 0.09%) and EGTA (final concentration 3 mmol/L), respectively[13]. Each tested compound was added to the cell suspension when the fluorescence intensity reached the stable state. The measurement of [Ca2+]i using Fura-2 is well established in our laboratory[13,24]. For every tested com-pound, we have observed its effect alone on [Ca2+]i for up to 15 min and can exclude non-specific effects due to the leakage of Fura-2 dye from cells. All chemicals were purchased from Sigma (USA).
Probe generation Human IL-2 and glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) cDNA was ligated into pBluescript II SK (+/-) vectors (Promega, USA), which contain the T3 and T7 RNA promoters. The plasmid DNA was isolated and linearized with KpnI or XbaI. Then [32P-CTP] (Dupont, USA)-labeled hIL-2 and human GAPDH riboprobes were synthesized by using linearized DNA templates, T3 RNA polymerase and the Riboprobe in vitro transcription system (Promega, USA). After extraction with phenol/chloroform and chloroform, and precipitation with ethanol, dried probes were dissolved (3×105 cpm/mL) in hybridization buffer.
RPA and RT-PCR analysis Purified human T cells (5×106 cells/mL) were incubated with different blockers for 15 min, at 37 °C, in 5% CO2, and then treated with 5 µg/mL ConA for 6 h. Total RNA was extracted using the acid guanidium thiocyanate-phenol-chloroform method[25].
The RPA was performed according to the manual supplied with the RNase protection kit (Ambion, USA). Briefly, a 15 µg total RNA fraction of each sample was hybridized with 1 µL labeled hIL-2 and human GAPDH riboprobes. The hybridized fragments protected from RNase A + T1 digestion were separated by electrophoresis on a standard 8% acrylamide/7M urea sequencing gel. Dried gels were placed on X-ray film for 24 h at -70 °C. The bands on the X-ray film were quantified by densitometry scanning (Koutron IBAS 2.0). The expression level of hIL-2 mRNA was expressed as the ratio of hIL-2/GAPDH.
For RT-PCR, 1 µg of the extracted RNA was used for synthesis of the first-strand cDNA using AMV reverse transcriptase (Promega) according to the manufacturer’s instructions. PCR amplification was carried out with 100 ng DNA in a total volume of 50 µL in a Gene Amp 9600 PCR system (Perkin-Elmer, Foster City, CA) for 25 cycles at 94 °C for 2 min, 55 °C (ClC-3) or 57 °C (β-actin) for 1 min, and extension at 72 °C for 1 min. The primers were 5'-ATTAAATGGATATA-CCCTTTCTTG-3' (sense) and 5'-TTGCAATTGTCAGGTC-TCTTCT-3' (antisense) for ClC-3[26]; 5'-GCTACAGCTTCA-CCACCAC-3' (sense) and 5'-TACTCCTGCTTGCTGA-TCCAC-3' (antisense) for β-actin[27]. The amplified products were electrophoresed on 2% agarose gels alongside a molecular weight marker. The PCR products were 288 and 498 bp in size for ClC3 and β-actin, respectively. Because the primers for β-actin were designed to span two exons and an intron, RT-PCR with β-actin primers controlled for genomic DNA contamination in the source RNA. The linear range of the PCR amplification was determined by carrying out the PCR for varying number of cycles with a fixed quantity of RNA (1 µg), so that the expression levels of ClC-3 could be semi-quantified by densitometry scanning and expressed as the ratio of ClC-3/β-actin.
Statistical analysis Results are expressed as mean±SEM. The IC50 values were estimated in each experiment on the basis of a semi-logarithmic concentration-dependent curve. Data were analyzed with the t-test using SPSS11. P<0.05 was considered significant.
Results
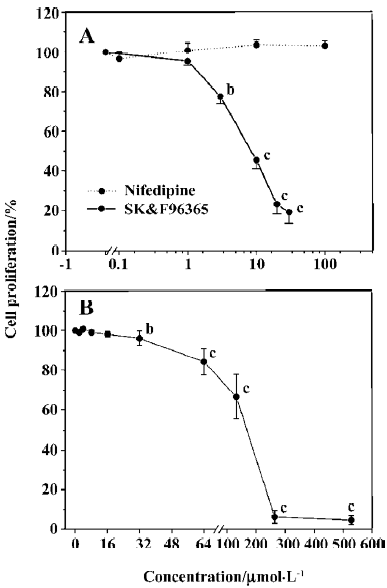
Effects of Ca2+ and Cl– channel blockers on ConA-induced T lymphocyte proliferation We first investigated whether CRAC and Cl– channel blockers affected T cell proliferation using an [3H]thymidine incorporation assay. As shown in Figure 1A, SK&F 96365, a common CRAC blocker, significantly inhibited cell proliferation at concentrations of 3 to 30 µmol/L in a concentration-dependent manner, with an IC50 value of 9.4±1.0 µmol/L, whereas nifedipine (0.1–100 µmol/L), an L-type VDCC inhibitor, did not significantly affect T lymphocyte proliferation (Figure 1A). These data indicate that Ca2+ entry through CRAC channels, but not VDCC, plays a functional role in T cell proliferation. Next we tested the effects of several Cl– channel blockers on human T cell proliferation. As shown in Figure 1B, DIDS significantly inhibited T cell proliferation at concentrations of 32 to 512 µmol/L in a concentration-dependent manner, with an IC50 value of 168.0±3.3 µmol/L, whereas no inhibitory effects on T cell proliferation were observed in the presence of 100 µmol/L 9-anthracene carboxylic acid (9-AC; n=5), 100 µmol/L niflumic acid (NFA; n=6) or 400 µmol/L furosemide (n=5; P>0.05, data not shown). Based on our previous findings that antisense oligonucleotides against ClC-3 decreased [3H]thymidine incorporation and prevented T cell proliferation[15], these data suggest that Cl– channels, which are sensitive to DIDS, may play a functional role during T lymphocyte proliferation.
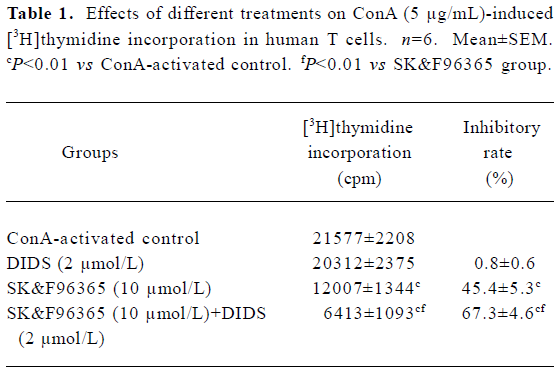
Because CRAC-mediated Ca2+ signaling is the predominant pathway during T cell activation and proliferation[2], the above results prompted us to further study whether Cl– channels would affect T cell proliferation through the CRAC-mediated Ca2+ signaling pathway. Although 2 µmol/L DIDS did not significantly affect cell proliferation, the inhibitory effect of SK&F96365 (10 µmol/L) on T cell proliferation was enhanced from 45.4%±5.3% (without DIDS) to 67.3%±4.6% (with 2 µmol/L DIDS; n=6, P<0.01; Table 1). These data suggest that Cl– channels may play a functional role in T cell proliferation through CRAC-mediated Ca2+ signaling.
Full table
Effects of Ca2+ and Cl– channel blockers on ConA-induced Ca2+ entry Measurement of [Ca2+]i using the Fura-2 probe revealed that the resting [Ca2+]i in human T cells was 67±5 nmol/L (n=54), which was not altered by depolarization of T cells with 50 mmol/L KCl (66±10 nmol/L, n=4, P>0.05), indicating a lack of VDCC in the T cell plasma membrane. We then examined the effects of 10 µg/mL ConA and 2 µmol/L TG on [Ca2+]i in human T cells, and representative traces are superimposed in Figure 2A. ConA (10 µg/mL) caused a transient and then a prolonged increase in [Ca2+]i, which was due to the corresponding release of Ca2+ from intracellular stores and the subsequent transmembrane Ca2+ entry, as shown in previous reports[3,5,22,28]. The increase in [Ca2+]i reached steady state within 150 s, and subsequent exposure of the cells to 2 µmol/L TG caused a further significant increase in [Ca2+]i (n=7). However, 10 µg/mL ConA failed to further increase Ca2+ influx in T cells whose stores were already depleted by 2 µmol/L TG (n=7). The pattern of the increase in [Ca2+]i stimulated by ConA was similar to that produced by 2 µmol/L TG except that the peak and the sustained plateau of [Ca2+]i caused by 10 µg/mL ConA were lower than those caused by 2 µmol/L TG (P<0.01, n=7; Figure 2B). These data indicate that ConA only evokes some Ca2+ release from TG-sensitive intracellular Ca2+ stores, which is enough to induce the Ca2+ influx responsible for T cell activa-tion and proliferation. Our data are consistent with the previous reports showing that lectin induces Ca2+ influx through the depletion of TG-sensitive intracellular Ca2+ stores[2,3,5,29]. The above proliferation assay demonstrated that 5 µg/mL ConA produced significant proliferative effects on T cells, but an even higher concentration of ConA (10 µg/mL) could not deplete the TG-sensitive intracellular Ca2+ stores completely; therefore, we used 5 µg/mL ConA as the stimulant for T cell activation in the subsequent experiments.
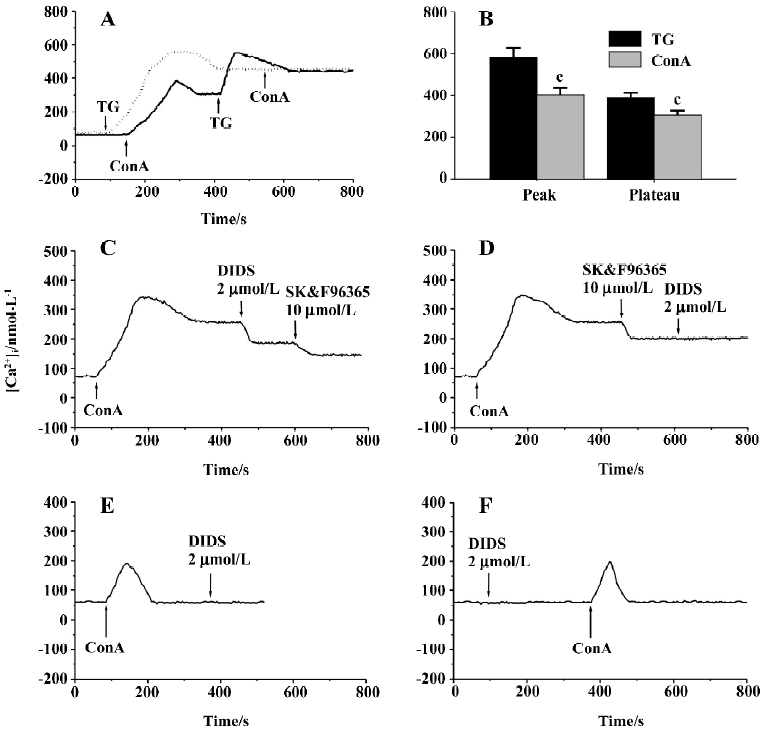
We next examined the effects of different [Ca2+]i channel blockers on 5 µg/mL ConA-induced Ca2+ entry. When Ca2+ entry reached a stable state, nifedipine was added to the cell suspensions, which did not affect the ConA-induced Ca2+ entry for concentrations of 0.1–10 µmol/L (Table 2), confirming that there are no VDCC in T cells, as described previously[2]. As shown in Table 2, SK&F96365 concentration-dependently inhibited ConA (5 µg/mL)-induced Ca2+ influx at concentrations of 1–20 µmol/L. At a concentration of 20 µmol/L, SK&F96365 inhibited Ca2+ influx by 52.9%±5.4%. SK&F96365 is an inhibitor of Ca2+ entry through CRAC channels, as well as through VDCC[23]. Because T cells do not contain VDCC, any effects of SK&F96365 should reflect its effects on CRAC channels. At concentrations of 0.5–2.0 µmol/L, DIDS also decreased Ca2+ influx in a concentration-dependent manner (Table 2). Because DIDS (>3 µmol/L) began to produce quenching effects on Fura-2 fluorescence, we did not test the effects of higher concentrations of DIDS. The results described so far suggest that DIDS affects [Ca2+]i. To investigate whether the inhibition of Ca2+ influx by DIDS is mediated through CRAC channels, the following pharmacological approaches were used. As shown in Figure 2C, 2 µmol/L DIDS caused a 32.3%±5.1% decrease in ConA (5 µg/mL)-induced Ca2+ influx (n=7, P<0.01), and subsequent exposure of cells to 10 µmol/L SK&F96365 further decreased Ca2+ influx by 19.6%±5.2% (n=7, P<0.01). However, when 10 µmol/L SK&F96365 caused a decrease in Ca2+ influx by 35.8%±6.2%(n=7, P<0.01), subsequent addition of 2 µmol/L DIDS brought about no change in [Ca2+]i (n=7, P>0.05; Figure 2D). Within the concentration range of 1–20 µmol/L, SK&F96365 serves as a CRAC channel blocker in T cells[2,4,23], so when the CRAC-mediated Ca2+ influx was prevented by 10 µmol/L SK&F96365, 2 µmol/L DIDS did not induce any change in [Ca2+]i, suggesting that DIDS inhibits the ConA-induced Ca2+ influx through interfering with the CRAC-mediated pathway. We also tested whether DIDS directly affects ConA (5 µg/mL)-induced Ca2+ release. Figure 2E shows that in Ca2+-free solution (following the removal of CaCl2 and after addition of 200 µmol/L EGTA), ConA (5 µg/mL) evoked a transient increase in [Ca2+]i, which was due to Ca2+ release from intracellular Ca2+ stores, but the subsequent application of 2 µmol/L DIDS failed to induce any change in [Ca2+]i (n=6, P>0.05; Figure 2E). In addition, pretreatment of cells with 2 µmol/L DIDS did not induce any change in [Ca2+]i in Ca2+-free solution and further failed to alter subsequent ConA-induced Ca2+ release (n=5, P>0.05, Figure 2F). These data indicate that 2 µmol/L DIDS had no direct effect on ConA-induced Ca2+ release or resting [Ca2+]i.

Full table
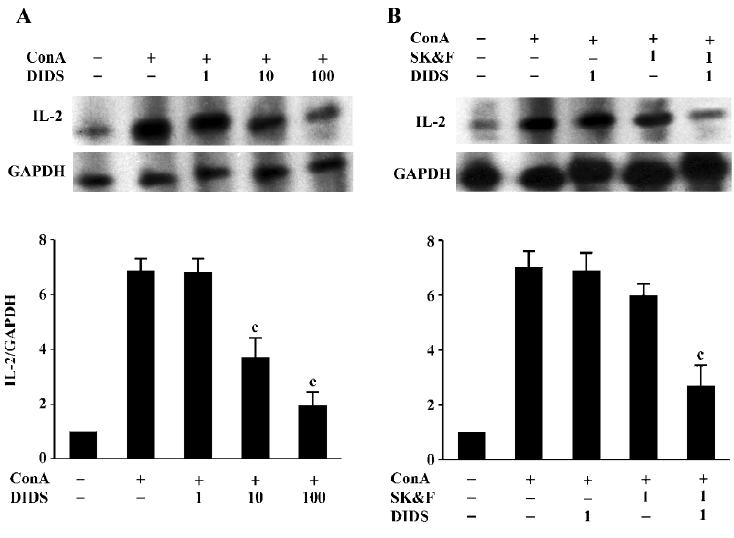
Effects of Ca2+ and Cl– channel blockers on ConA-induced IL-2 mRNA expression In T cells, CRAC-mediated sustained increase in [Ca2+]i activates several transcription factors, including NF-κB, Oct/OAP, and NF-AT, then initiates the transcription of IL-2 mRNA, which results in IL-2 production and finally proliferation of T cells[2]. RPA analysis demonstrated that ConA (5 µg/mL) increased IL-2 mRNA expression in human T cells after 6 h of incubation (662%±45% increase; n=7, P<0.01). To examine the effects of ion channel blockers on ConA-stimulated IL-2 mRNA expression, cells were incubated with the blockers for 30 min prior to exposure to ConA. As shown in Figure 3A, 10 and 100 µmol/L DIDS caused a decrease in ConA-stimulated hIL-2 mRNA expres-sion, with inhibitory rates of 37%±8% and 69%±7%, respectively (n=4, P<0.01 vs ConA-stimulation control), whereas 1 µmol/L DIDS had no effect on IL-2 mRNA expression in stimulated T cells (n=4, P>0.05 vs ConA-stimulation control). Consistent with previous reports[4], SK&F96365 also decreased IL-2 mRNA expression. We observed that 1 µmol/L SK&F96365 decreased IL-2 mRNA expression by 12%±5% (n=5, P>0.05 vs ConA-treatment group; Figure 3B). The inhibition of ConA-stimulated hIL-2 mRNA expression was significantly enhanced by the combination of 1 μmol/L SK&F96365 and 1 µmol/L DIDS (n=5, P<0.01 vs the corresponding ConA plus blocker group). The inhibitory rates for DIDS, SK&F96365, and SK&F96365 plus DIDS were 2%±1%, 12%±5%, and 66%±9%, respectively. It has been demonstrated that naive T cell stimulation induces IL-2 mRNA, which is present for at least 5 h. This short burst of IL-2 mRNA allows for the secretion of functional levels of IL-2 protein that are sufficient to induce naive T cells to enter the cell cycle[30]. In the present study, we harvested total mRNA after the co-incubation of cells with the tested compounds for 6 h. Thus, comparing the effects of both channel blockers on IL-2 mRNA expression could reflect the functional role of the Cl– channels during Ca2+-dependent T cell proliferation signaling cascades.
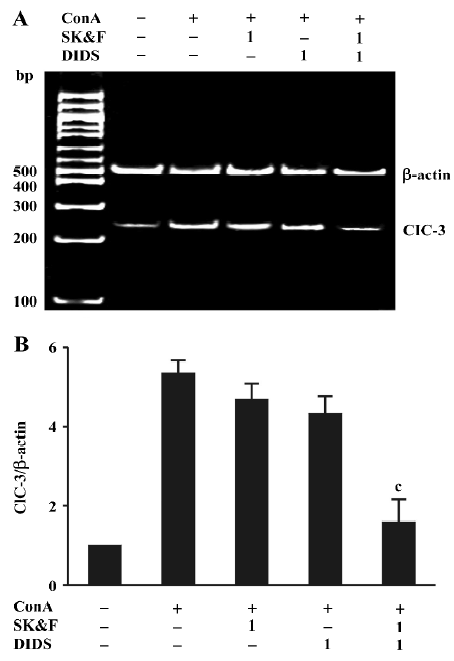
Effects of Ca2+ and Cl- channel blockers on ConA-induced ClC-3 expression Our recent studies support the notion that ClC-3 may be the potential functional player during T cell proliferation[15,16]. Therefore, we examined the effects of DIDS and its synergistic effect together with SK&F96365 on ClC-3 mRNA expression. As shown in Figure 4A, using primers specific to the human cDNA sequence of ClC-3, bands corresponding to the expected fragment size (248 bp) were obtained from the total RNA prepared from human T lymphocytes exposed to different treatments. ClC-3 transcripts were found in all samples, and were increased by ConA-treatment, which was consistent with the ClC-3 protein expression described in human T lymphocytes using Western blot analysis[16]. As summarized in Figure 4B using ClC-3/β-actin ratios, the exposure of T cells to 1 μmol/L DIDS and 1 µmol/L SK&F96365 decreased ConA-induced ClC-3 mRNA expression by 16%±6% and 9%±4%, respectively (n=6, P>0.05 vs ConA-treatment group). Moreover, the combination of 1 µmol/L DIDS and 1 µmol/L SK&F96365 acted synergistically to diminish ClC-3 mRNA expression by 64%±10% (n=6, P<0.05 vs the corresponding ConA plus blocker group). The β-actin bands corresponding to 500 bp are the same in all lanes. These data suggest that the effect of DIDS on the ConA-induced ClC3 message may be linked to CRAC-mediated signaling during T cell activation.
Discussion
The present study demonstrates the following events: (1) DIDS, a chloride channel blocker, inhibits ConA-induced human T lymphocyte proliferation in a concentration-dependent manner, but 9-AC, NFA, and furosemide have no significant effect on T cell proliferation induced by ConA; (2) when combined with SK&F96365, DIDS enhances the inhibitory effect of SK&F96365 on ConA-induced IL-2 mRNA expression and T cell proliferation; (3) both DIDS and SK&F96365 decreased ConA-induced Ca2+ influx, but the mechanisms used by the two inhibitors may be different; (4) a combination of DIDS and SK&F96365 enhanced the inhibition of ConA-induced ClC-3 mRNA expression relative to the effect of each alone.
Fura-2 fluorescence analysis showed that ConA-treatment did not further increase Ca2+-influx in T cells whose stores had been already depleted by TG. This observation agrees with the recognized idea that CRAC channel is solely responsible for Ca2+-influx during lectin-induced T cell activation[2]. Thus, the effects of SK&F96365 on ConA-induced T cell activation and proliferation can be attributed to the inhibition of Ca2+-influx through CRAC channel.
A growing number of reports suggest that Cl– channels play a functional role in the cellular proliferation process in T cells and other cell types[12,13,17,19], which was further supported by our present work examining the effects of DIDS on several pivotal steps during T cell proliferation, including early Ca2+ signaling and IL-2 mRNA expression as well as later cell proliferation. In our present and previous studies, DIDS has been shown to prevent cell proliferation in T lymphocytes stimulated by lectins (ConA) and in rat aortic smooth muscle cells stimulated by ET-1[13,18]. However, it should be noted that DIDS is also an effective blocker of Cl–/HCO3– exchangers, ATP-sensitive K+ channels, and can bind to other proteins[31,32]. Due to the lack of specific chloride channel blockers, it is difficult to confirm whether Cl– channels play a functional role in cell proliferation just using Cl– channel blockers. To exclude several ion transports from the potential candidate list for cell proliferation, we first compared the effects of chemically different Cl– channel blockers on ConA-induced T lymphocyte proliferation. As shown in our previous studies[13,18], it appears that the effect of DIDS and NPPB on proliferation is not related to Ca2+-activated or maxi Ca2+ channels, because the Cl– channel inhibitors NFA (Ca2+-activated Cl– channel blocker), SITS (Ca2+-activated and maxi Cl– channel blocker), and IAA-94 (Ca2+-activated Cl– channel blocker) did not change the effect of ConA on T cell proliferation. Among the ClC family members identified in immune cells (ClC-2, 3, 4, 5), ClC-2 has been excluded from the candidate lists for cellular proliferation[17]. Another potential candidate may be the ClC-3 Cl– channel. Abundant expression of ClC-3 mRNA and protein has been detected in T lymphocytes[16,26]. Our previous work provided compelling evidence of the functional role of ClC-3 protein in ConA-induced T cell proliferation using antisense oligonucleotides against ClC-3. We found that the downregulation of ClC-3 by antisense, but neither sense or missense, oligonucleotides against ClC-3 not only inhibited ConA-induced ClC-3 expression but also decreased [3H]thymidine incorporation and prevented T cell proliferation[15,16]. The Cl– current recorded in T cells shares similar pharmacological properties with the expressed ClC-3 currents in mammalian cell lines, such as sensitivity to Cl– channel blockers including DIDS, NPPB and tamoxifen[33,34]. These studies, along with the present work examining the effects of Cl– channel blockers on several key signaling steps during T cell proliferation, strongly suggest that ClC-3 channels may play functional roles during T cell proliferation signaling cascades. Although it is still unclear whether the ClC-3 protein is a volume-regulated channel or a component thereof in some cell types[35,36], the involvement of ClC-3 protein in cell proliferation has received strong support from studies carried out in our laboratory and other independent studies[12]. On the other hand, the present study could not indicate whether ClC-3 is solely responsible for Cl– channel activation by ConA in T cells. We have not accepted Ca2+-activated Cl– channels (ICl(Ca)) as potential candidates, because NFA, a very common blocker of ICl(Ca) did not alter T cell activation. Elucidating the exact nature of Cl– channel activation in T cell proliferation will require further study.
In addition to their direct effect on T cell activation, Cl– channels may participate in T cell activation through the regulation of Ca2+ signaling. Although CRAC channels are not opened directly by depolarization, Ca2+ ions entering the cell through CRAC channels feed back to inhibit the channel’s activity by reducing the membrane potential. K+ and Cl– channels have been found to assist with Ca2+ entry by maintaining a sufficiently negative membrane potential[37]. Therefore, it is reasonable to suggest that Cl– channels may be involved in T cell activation by regulation of the CRAC channels. Our data also indicated that ConA-induced Ca2+ release was not significantly changed by DIDS, which further supports the finding that DIDS at low concentrations has no effect on Ca2+ release in some cell types[38,39]. Thus, it seems that the effect of DIDS on T cell activation is due to the inhibition of CRAC-induced Ca2+ entry. This appears to be the first description at different molecular levels of the interaction between Cl- channels and CRAC-mediated Ca2+ signaling during T cell activation. Although the link between Cl– channels and CRAC-mediated Ca2+ influx cannot be fully explained by the quenching effects of DIDS (>3 µmol/L) on Fura-2 fluorescence, the concentration range of DIDS that produced effects on ClC-3, IL-2 mRNA expression and cell proliferation agree with the characterization of Cl- channels determined in other studies[19,28,34,40].
Although a sustained Ca2+ plateau is the critical trigger inducing downstream IL-2 gene expression and cell proliferation in ConA-activated T cells, how the specific information embedded in the amplitude, duration and kinetics of Ca2+ signals is transmitted to downstream selective gene expression in T cells remains an unanswered key question in cell signaling[8]. In addition, the various sizes of commonly distributing Ca2+ oscillations also adds more complexity to Ca2+ signaling in T cells[8]. Therefore, T lymphocyte Ca2+ signals are more than a binary switch in T cell activation, and different patterns of Ca2+ signals may transmit according to the specific patterns of gene expression, which will elicit final selective responses in activated T cells. The emerging complex Ca2+ signaling in T cell activation may also help to explain the inconsistencies in effective DIDS concentrations in the present study. A concentration of 2 µmol/L DIDS significantly inhibited CRAC-induced Ca2+ entry, but had no effect on IL-2 mRNA expression and final T cell proliferation. It is reasonable to assume that DIDS-sensitive Cl– channels play a time-dependent functional role during the whole process of cellular proliferation. Acute exposure to DIDS (less than a few seconds) at a low concentration may only transiently reduce Ca2+ signaling and fail to affect later gene expression and final cell proliferation; whereas long-term exposure (up to a number of hours) to higher concentrations of DIDS may cause complete inhibition of specific Ca2+ signaling, leading to the inhibition of specific gene expression and final T cell proliferation.
In summary, the results of our present study suggest that DIDS-sensitive Cl– channels may play a functional role in ConA-stimulated, Ca2+-dependent IL-2 mRNA expression and cell proliferation in T cells through CRAC-mediated Ca2+ entry. Definitive molecular determination of the DIDS-sensitive Cl– channels will require gene targeting experiments combined with more specific pharmacological tools. ClC-3 may be responsible for the DIDS-sensitive Cl– channel, which may serve as a novel interfering target during the signaling transduction of T cell activation and proliferation.
Footnote
Abbreviations used in this paper: ConA, concanavalin A; [Ca2+]i, intracellular free calcium concentration; CRAC, Ca2+-release-activated Ca2+; DIDS, 4,4'-diisthiocyanostilbene-2,2'-disulphonic acid; SK&F96365, 1-{beta-[3-(4-methoxyphenyl)propoxyl]-4-methoxy-phenethyl}-1H-imidazole; TG, thapsigargin; NPPB, 5-nitro-2-(5-phenylpropylamino)-benzoate; 9-AC, 9-anthracene carboxylic acid; NFA, niflumic acid; STIS, 4-acetamido-4-isocyana-tostilbene-2,2-disulphonic acid;IAA-94, Indanyloxyacetic acid; RPA, RNase protection assay; VDCC, voltage-dependent Ca2+ channel.
References
- Gardner P. Calcium and T lymphocyte activation. Cell 1989;59:15-20.
- Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol 2001;19:497-521.
- Premack BA, McDonald TV, Gardner P. Activation of Ca2+ current in Jurkat T cells following the depletion of Ca2+ stores by microsomal Ca2+-ATPase inhibitors. J Immunol 1994;152:5226-40.
- Chung SC, McDonald TV, Gardner P. Inhibition by SK&F96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br J Pharmacol 1994;113:861-8.
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA 1993;90:6295-9.
- Sarkadi B, Tordai A, Homolya L, Scharff O, Gardos G. Calcium influx and intracellular calcium release in anti-CD3 antibody-stimulated and thapsigargin-treated human T lymphoblasts. J Membr Biol 1991;123:9-21.
- Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immuno-deficiency. J Biol Chem 1994;269:32327-35.
- Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol 1995;131:655-67.
- Rouzaire-Dubois B, Milandri JB, Bostel S, Dubois JM. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch 2000;440:881-8.
- Voets T, Szucs G, Droogmans G, Nilius B. Blockers of volume-activated Cl- currents inhibit endothelial cell proliferation. Pflugers Arch 1995;431:132-4.
- Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, et al. Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J Physiol 2001;532:661-72.
- Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY. Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res 2002;91:E28-32.
- Xiao GN, Guan YY, He H. Effects of Cl– channel blockers on endothelin-1-induced proliferation of rat vascular smooth muscle cells. Life Sci 2002;70:2233-41.
- Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol 2005;145:5-14.
- Qian Y, Guan YY, Wang GL, Yang XR, He H, Qiu QY. Effects of ClC-3 antisense oligonucleotide on ConA induced T lymphocyte proliferation. Chin Pharmacol Bull 2004;20:314-7.
- Qian Y, Guan YY, Wang GL, Yang XR, He H, Qiu QY. Relationship between ConA induced T lymphocyte proliferation and ClC-3 protein. Chin Pharmacol Bull 2003;19:1132-5.
- Jiang B, Hattori N, Liu B, Nakayama Y, Kitagawa K, Inagaki C. Suppression of cell proliferation with induction of p21 by Cl- channel blockers in human leukemic cells. Eur J Pharmacol 2004;488:27-34.
- Wang GL, Guan YY, Wang B. Effects of chloride-channel blockers on T lymphocyte proliferation. Chin Pharmacol Bull 1999;15:146-9.
- Phipps DJ, Branch DR, Schlichter LC. Chloride-channel block inhibits T lymphocyte activation and signalling. Cell Signal 1996;8:141-9.
- Hess SD, Oortgiesen M, Cahalan MD. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J Immunol 1993;150:2620-33.
- Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol 2001;2:316-24.
- Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul 1989;1:99-112.
- Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, et al. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J 1990;271:515-22.
- Tao L, Guan YY, He H, Han C, Zhang YY, Sun JJ. Comparison of the Ca2+ movement by activation of alpha1-adrenoceptor subtypes in HEK-293 cells. Life Sci 1997;61:2127-36.
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156-9.
- Jiang B, Hattori N, Liu B, Kitagawa K, Inagaki C. Expression of swelling- and/or pH-regulated chloride channels (ClC-2, 3, 4 and 5) in human leukemic and normal immune cells. Life Sci 2002;70:1383-94.
- Britton FC, Ohya S, Horowitz B, Greenwood IA. Comparison of the properties of CLCA1 generated currents and ICl(Ca) in murine portal vein smooth muscle cells. J Physiol 2002;539:107-17.
- Lewis RS, Ross PE, Cahalan MD. Chloride channels activated by osmotic stress in T lymphocytes. J Gen Physiol 1993;101:801-26.
- Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem 1994;63:1045-83.
- Zuckerman LA, Pullen L, Miller J. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J Immunol 1998;160:3259-68.
- Takahashi N, Wang X, Tanabe S, Uramoto H, Jishage K, Uchida S, et al. ClC-3-independent sensitivity of apoptosis to Cl- channel blockers in mouse cardiomyocytes. Cell Physiol Biochem 2005;15:263-70.
- Zhang H, Zhou JG, Qiu QY, Ren JL, Guan YY. ClC-3 chloride channel prevents apoptosis induced by thapsigargin in PC12 cells. Apoptosis 2006;11. in press.
- Dietrich J, Lindau M. Chloride channels in mast cells: block by DIDS and role in exocytosis. J Gen Physiol 1994;104:1099-11.
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature 1997;390:417-21.
- Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, et al. ClC-3 is a fundamental molecular component of volume-sensitive outwardly rectifying Cl– channels and volume regulation in HeLa cells and Xenopus laevis oocytes. J Biol Chem 2002;277:40066-74.
- Sardini A, Amey JS, Weylandt KH, Nobles M, Valverde MA, Higgins CF. Cell volume regulation and swelling-activated chloride channels. Biochim Biophys Acta 2003;1618:153-62.
- Kerschbaum HH, Negulescu PA, Cahalan MD. Ion channels, Ca2+ signaling, and reporter gene expression in antigen-specific mouse T cells. J Immunol 1997;159:1628-38.
- Sipido KR, Callewaert G, Carmeliet E. [Ca2+]i transients and [Ca2+]i-dependent chloride current in single Purkinje cells from rabbit heart. J Physiol 1993;468:641-67.
- Verkerk AO, Schumacher CA, van Ginneken AC, Veldkamp MW, Ravesloot JH. Role of Ca2+-activted Cl- current in ventricular action potentials of sheep during adrenoceptor stimulation. Exp Physiol 2001;86:151-9.
- Schumacher PA, Sakellaropoulos G, Phipps DJ, Schlichter LC. Small-conductance chloride channels in human peripheral T lymphocytes. J Membr Biol 1995;145:217-32.