
All-trans retinoic acid inhibited angiotensin II-induced increase in cell growth and collagen secretion of neonatal cardiac fibroblasts1
Introduction
Increased hemodynamic workload is well known to alter the structure and muscle content of the heart[1,2]. These responses, as component parts of cardiac remodeling, are thought to be an adaptive mechanism to decrease cardiac wall stress and maintain heart function. However, cardiac remodeling also results in increased myocardial stiffness and ventricular diastolic dysfunction, potentially leading to chronic heart failure. This is an important cause of morbidity and mortality associated with cardiovascular disease.
Cardiac remodeling is the composite result of many factors, including mechanical stretching, ischemia, and the effects of hormones and cytokines. During cardiac remodeling, cardiac fibroblasts (CF) undergo significant changes: they proliferate and produce an increased amount of extracellular matrix, including fibronectin, laminin and collagen types I and III. In addition, they may assume a myofibroblast character and express α-actin, and thus contain myofilaments[3–7]. These changes may lead to fibrous material deposition, alterations of the highly organized extracellular matrix and interconnections between cardio-myocytes and their neighboring cells, thus impairing the systolic and diastolic functions of the heart.
Angiotensin II (Ang II) is a major final effector molecule of the renin-angiotensin system. Ang II may act as a co-mitogen for the proliferation of some types of cells. It has also been implicated as an important factor that contributes to the pathogenesis of atherosclerosis, hypertension, and cardiac remodeling[4,5,8–10]. Ang II is also known to stimulate cell proliferation and secretion of types I and III in CF, and activate the expression of some growth factors, such as the transforming growth factor-β1 (TGF-β1)[6], which in turn modulates the cell’s response to Ang II. Therefore, Ang II plays an important role in cardiac fibrosis. In this context, drugs that decrease or antagonize the effect of Ang II may be beneficial for alleviating cardiac remodeling and heart failure.
All-trans retinoic acid (atRA), a derivative of vitamin A, influences cellular differentiation in organ development[11,12], and inhibits proliferation of a variety of cell types[12–14]. WANG et al determined that atRA inhibited Ang II-induced cardiomyocyte hypertrophy[15], and LÜ et al demonstrated that chronic treatment with atRA prevented medial thickening of intramyocardial and intrarenal arteries, and ventricular fibrosis during the development of hypertension in spontaneously hypertensive rats[16]. There is evidence that retinoids reduce the expression of platelet-derived growth factor, inducible nitric oxide synthase, interleukin-6, and endothelin[12,13,17], thus opposing the action of Ang II. The effects of atRA on the expression and function of TGF-β1 as well as extracellular matrix proteins are cell specific[18,19]. It has been reported that atRA blocks the stimulatory action of Ang II on TGF-β1 mRNA in vascular smooth muscle cells[20]. Therefore, it is interesting to ascertain whether atRA has similar effects on CF. The purpose of the present study was to determine whether atRA alleviated the Ang II-induced increase in cell growth and collagen secretion in cultured CF from neonatal rats.
Materials and methods
CF cell culture CF were isolated from 1-day-old Sprague-Dawley rat pups with 0.1% trypsin. To select pure cardiac fibroblasts, dissociated cells were preplated in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Rockville, MD, USA) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA) at 37 °C with 5% CO2 for 1 h. The nonmyocytes attached readily to the bottom of the culture dish. Cells adhered to the culture dishes during the preplating procedure were resuspended with culture medium and then allowed to reattach for 30 min at 37 °C. After this procedure was repeated twice, the cells were distributed into culture dishes and incubated in the culture medium containing 10% fetal bovine serum. In the third passage, the cells were mainly CF. Cells from the third or fourth passages were used for experiments.
Cell growth assay Subconfluent CF were seeded into 96-well plates at a density of 4000 cells per well and cultured for 12 h in the presence of 10% fetal bovine serum (Gibco). Subsequently, cells were washed twice with serum-free medium and growth was arrested in serum-free medium for 24 h. Then the quiescent cells were treated with Ang II and/or atRA for 24 h. More Ang II was added after 12 h to compensate for a decrease due to degradation by endogenous angiotensinase. Control cultures contained dimethyl sulfoxide (Me2SO; Sigma, St Louis, MO, USA). After incubation of the cultures for 24 h at 37 °C, 10−20 µL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma) was added per well. The culture plate was preplated in a cabinet at 37 °C with 5% CO2 for 4 h, then the old medium was removed and 150 µL Me2SO was added per well. The culture plate was shaken for 15 min. Absorbence at 490 nm was considered to be directly proportional to the number of cells in the wells and was therefore used as a measure of cell growth.
Immunocytochemistry analysis CF cultured on 12-mm glass coverslips were serum-starved for 24 h and then cultured for 48 h in media alone (control) or treated with Ang II and/or atRA. CF were then fixed in 10% buffered formalin for 10–30 min, washed twice with distilled water, and immersed in 30% H2O2/carbinol for 30 min at room temperature. After that, the cells were washed 3 times with distilled water, incubated with 5% bovine serum albumin (BSA; Boster, Wuhan, China) for 20 min, then with collagen type I or collagen type III antibodies (1:50) (Boster) for 12 h at 4 °C. After 3 washes with phosphate-buffered saline (PBS) and incubation with horseradish peroxidase-conjugated goat anti-mouse antibody (1:2000; Boster) for 20 min at 37 °C, CF were washed again with PBS, then incubated with streptavidin-biotin-peroxidase complex (SABC, Boster) for 20 min. After being washed for 20 min with PBS by using a DAB-kit (Boster), CF were mounted on cover slides for microscopic imaging.
Western blot analysis The protein concentrations were determined by using a BCA-100 protein quantitative analysis kit (Shenergy Biocolors). An equal amount of protein from the medium was subjected to 5%–12% gradient sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Amersham Pharmacia). After electrophoretic separa-tion, proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Amersham Pharmacia), which was then incubated overnight at 4 °C with the primary antibody (1:500; Santa Cruz, Santa Cruz, USA). Antibody binding was detected by counter-staining with horseradish peroxidase-conjugated antibodies (1:2000; CalBiochem, USA) and visualized using an ECL-detection kit (Pierce Biotechnology, Rockford, IL, USA). The efficiency of transfer was confirmed by staining the membrane with Ponceau S. The relative intensity of immunoreactive bands on the exposed film was quantified by using a computer-assisted densitometry program (Smart View; Life Science Research Products and System Engineering).
Statistical analysis Data are presented as mean±SD. Statistical significance was evaluated by one-way ANOVA followed by the Turkey post hoc test (SPSS software). Significance was accepted at P<0.05. Each experiment was repeated 3 times to verify the reproducibility of the results.
Results
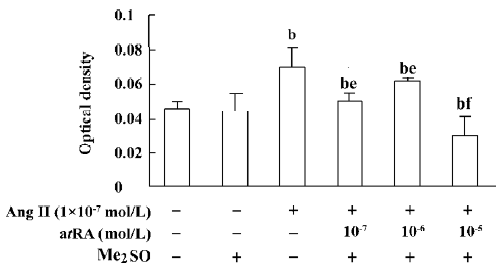
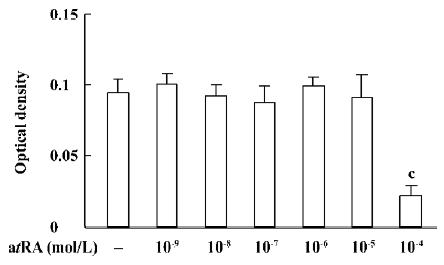
atRA attenuated the Ang II-induced increase in cell growth of CF Exposure of neonatal rat CF for 24 h to Ang II (1×10-9 to 1×10-6 mol/L) induced a significant increase in cell growth, therefore Ang II at a concentration of 1×10-7 mol/L was used in the subsequent experiments. The Ang II-induced increase in cell growth was attenuated by atRA (1×10-7 to 1×10-5 mol/L). When CF were co-treated with atRA (1×10-5 mol/L) and Ang II for 24 h, the cell growth level was even below that of the control (Figure 1). atRA alone at concentrations of 1×10-9 to 1×10-5 mol/L produced no effect on the cell growth of CF, but caused a significant decrease at a higher concentration (1××10-4 mol/L; Figure 2).
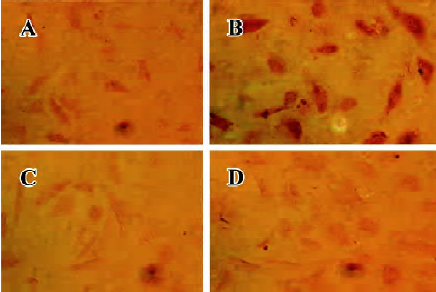
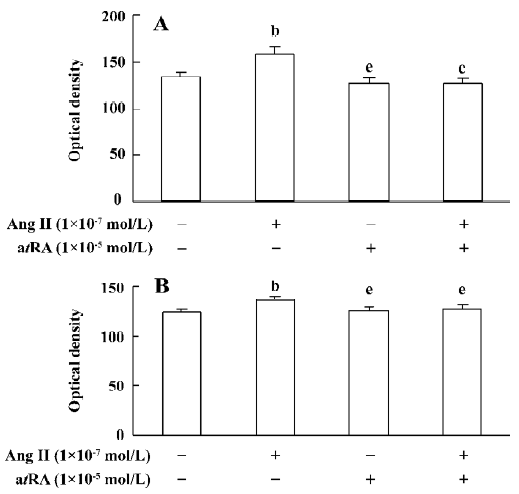
atRA attenuated the Ang II-induced increase in intracellular collagen types I and III in CF Immunocytochemical staining was used to detect intracellular collagen types I and III. In the control neonatal rat CF, only sparsely distributed collagen type III staining was observed (Figure 3A). Exposure to Ang II (1×10-7 mol/L) for 48 h resulted in an accumulation of collagen type III in CF (Figure 3B). atRA (1×10-5 mol/L) treatment reduced the Ang II-induced accumulation of collagen type III (Figure 3D). atRA by itself (1×10-5 mol/L) caused no apparent change in the quantity of collagen type III (Figure 3C). The results of image analysis are shown in Figure 4A. Similarly, the Ang II-induced increase in the content of collagen type I in the CF was also abolished by atRA (Figure 4B).
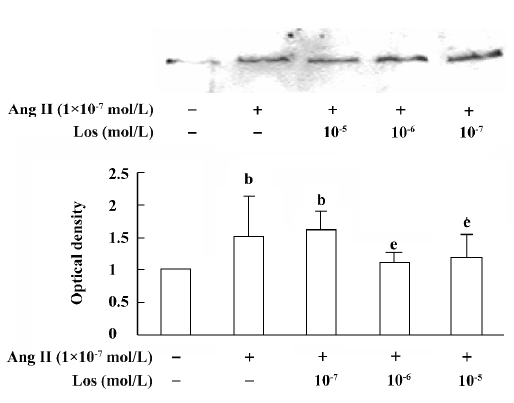
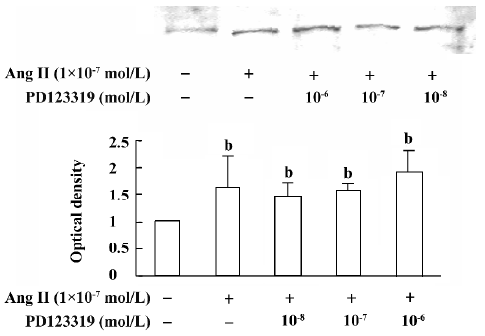
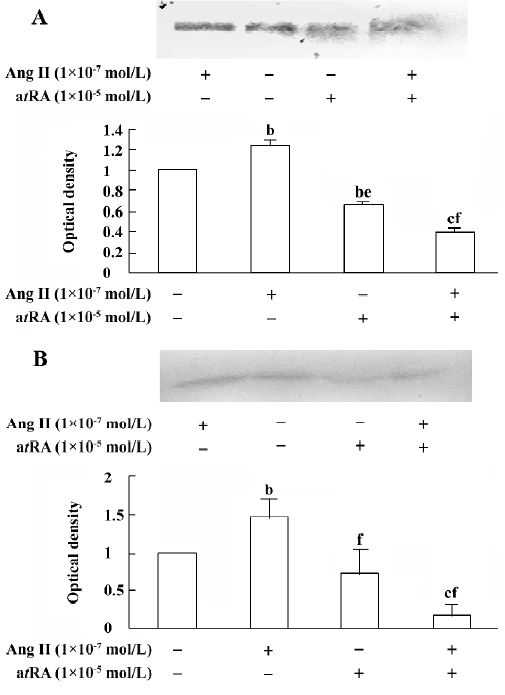
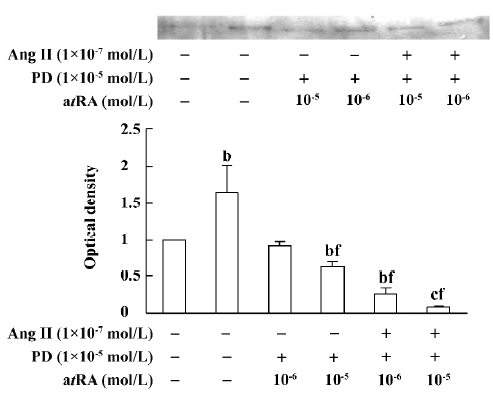
atRA attenuated the Ang II-induced increase in collagen secretion of CF Western blot analysis was used to measure collagen secretion by the neonatal rat CF into the culture medium. Figure 5 shows that an increase in collagen type III in the culture medium was detected in Ang II (1×10-7 mol/L)-treated cultures and that this response was blocked by angiotensin AT1 receptor antagonist losartan (1×10-6 and 1×10-5 mol/L). However, this effect of Ang II was not affected by angiotensin AT2 receptor antagonist PD123319 (up to 1×10-6 mol/L; Figure 6). atRA (1×10-5 mol/L) alone slightly reduced the basal secretion of collagen types III and I, and completely abolished the Ang II-induced stimulation of collagen secretion (Figure 7A,7B). When the CF were co-treated with atRA and Ang II, the collagen levels in the culture medium were significantly lower than those of the controls (Figure 7). We also observed that when PD123319 (1×10-5 mol/L) was added to the culture medium, application of atRA (1×10-6 or 1×10-5 mol/L) together with Ang II (1×10-7 mol/L) again significantly reduced the secretion of collagen type III into the medium (Figure 8).
atRA attenuated the Ang II-induced increase in TGF-β1 expression in CF Western blot analysis was used to measure the expression of TGF-β1 in CF. After 48 h of exposure to Ang II (1×10-7 mol/L), the expression of TGF-β1 in the CF was significantly increased compared with the controls. atRA (1×10-5 mol/L) alone caused no obvious change in the basal level of TGF-β1 in the CF, but it completely abolished the Ang II-induced stimulation of TGF-β1, the TGF-β1 content was significantly lower than that of the control (Figure 9).
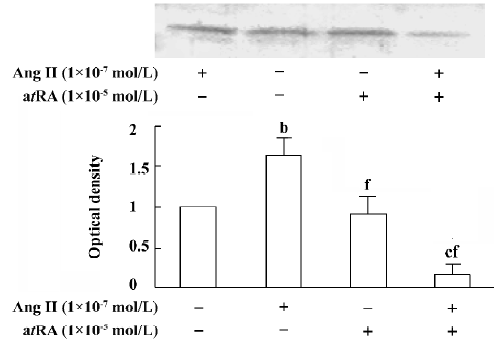
Discussion
Our observations demonstrate that atRA inhibits the stimulatory effect of Ang II on cultured neonatal rat CF, which is consistent with the results previously achieved in our laboratory by measuring the myocyte protein content[15]. In the present study, we also found that atRA (up to 1×10-5 mol/L) by itself generally produced no apparent effect on CF cell growth, but a high concentration of atRA (1×10-4 mol/L) caused a significant decrease, possibly indicating toxicity. Therefore, atRA at concentrations of 1×10-5 and 1×10-6 mol/L were used in the present study to observe its effect on the actions of Ang II.
The induction of growth and proliferation of a variety of cell types by Ang II has been intensively studied. The cell response to Ang II depends on the interactions between pro- and anti-proliferative factors, and it is well known that Ang II stimulates the expression of a number of pro-proliferative autocrine factors. Ang II has been demonstrated to be closely associated with the production of extracellular matrix. An accumulation of fibrillar collagen in the interstitial space of the hypertrophied heart is thought to be responsible for abnormal ventricular wall stiffness and for impaired cardiac pumping capacity[21,22]. It has been shown that Ang II increases cardiac fibroblast-mediated collagen synthesis and mRNA levels of collagen types I and III[23]. The immunocytochemistry results in the present study showed that 48 h exposure of neonatal rat CF to Ang II resulted in an increase in the collagen types I and III content. Because collagen is secreted into the extracellular matrix as soon as it is synthesiz-ed, we measured the collagen content in the culture medium, and our results from Western blotting showed that the secretion of collagen types I and III was stimulated by Ang II. We also demonstrated that the stimulatory action of Ang II was completely blocked by the angiotensin AT1, but not AT2, receptor antagonist, indicating that the Ang II-induced increase in collagen secretion was mediated mainly by activating angiotensin AT1 receptors.
It has been documented that atRA promotes differentiation and fibrinolysis, but also inhibits cell proliferation, migration, thrombosis, angiogenesis, platelet aggregation, and inflammation[24–27]. In line with the finding that atRA attenuated the effect of Ang II on cell growth, we also found that atRA inhibited the Ang II-induced increase in secretion of collagen types I and III by the cultured neonatal rat CF.
TGF-β1 is an important growth factor involved in the development of myocardial fibrosis. Low expression levels of TGF-β1 and collagen types I and III mRNA are seen in the normal rat heart; however, expression of TGF-β1 and collagen types I and III is markedly increased in the infarcted heart. The increase in TGF-β1 mRNA precedes the increases in collagen mRNA levels. In in vitro studies, TGF-β1 has been found to induce an increase in collagen production and secretion, and enhance the mRNA levels of collagen types I and III in rat CF[28]. Increased myocardial TGF-β1 and collagen mRNA are found in myocardial fibrosis induced by Ang II infusion[28]. There is also evidence indicating that TGF-β1 inhibits the production and secretion of collagenase (MMP-1), leading to the accumulation of collagen in the extracellular matrix[29]. Our present results have revealed that Ang II produces a significant activation of TGF-β1 in cultured neonatal rat CF, and that this effect of Ang II is abolished by atRA. Combined administration of atRA and Ang II caused a significant decrease in TGF-β1 below the basal level.
The mechanism underlying the effect of atRA on Ang II-induced activation of CF remains to be further clarified. In the present study, we did not measure the expression levels of AT1 and AT2 receptors in atRA-treated CF; however, there is evidence that atRA downregulates AT1 receptor levels in vascular smooth muscle cells[20]. This might be also true in CF. Because the Ang II-induced activation of CF is mediated via the AT1 receptor, it is possible that atRA inhibits the effect of Ang II on CF via downregulation of the AT1 receptor. An interesting observation in the present study is that combined treatment with atRA and Ang II resulted in a significant decrease in the secretion of collagen and the expression of TGF-β1 below their respective basal levels. It is known that activation of angiotensin AT2 receptor produces effects that are opposite to those of AT1 receptor activation, so a possible explanation is that when cells are treated with both atRA and Ang II, the AT1 receptor-mediated effect is blocked or attenuated by atRA, whereas the AT2 receptor-mediated effect becomes apparent, leading to decreases in TGF-β1 expression and collagen secretion. However, in the present study we found that when AT2 receptor antagonist PD123319 was administered together with atRA and Ang II, the secretion of collagen type III was as low as that without PD123319 administration. We therefore suggest that the inhibition of collagen secretion upon combined treatment of the CF with atRA and Ang II is not mediated by activation of AT2 receptors. Combined treatment with atRA and Ang II probably results in activation of some other signals or signaling pathways, leading to a decrease in the secretion of collagen. Because TGF-β1 is known to inhibit the expression of collagenase, and in the present study we observed that the expression of TGF-β1 was significantly decreased when CF were co-treated with atRA and Ang II, we therefore hypothesize that combined administration of atRA and Ang II results in a decrease in the expression of TGF-β1 via some un-known pathway and, in turn, a low level of TGF-β1 leads to an increase in the activity of collagenase and thus decreases the collagen content. Our data are similar to those reported by Haxsen et al[20], who noted that combined administration of atRA (1 µmol/L) and Ang II (1 µmol/L) in a culture of vascular smooth muscle cells resulted in a significant decrease in the level of TGF-β1 mRNA. They suggested that the abrogation of Ang II-dependent TGF-β1 induction by atRA in vascular smooth muscle cells was possibly mediated via the activator protein-1 (AP-1)[20].
In summary, our observations provide further evidence that atRA inhibits Ang II-induced increases in cell growth and secretion of collagen types I and III in cultured neonatal rat CF. We hypothesize that this inhibition of collagen secretion might be related to the decrease of TGF-β1 expression in CF. These data support the notion that atRA exerts a beneficial effect on cardiac structural impairment during the process of cardiac remodeling. The details of the mechanism by which atRA acts remain to be further elucidated.
Acknowledgement
We would like to thank Mr Yin-xiang CAO for his valuable assistance with image analysis.
References
- Weber KT, Anversa P, Armstrong PW. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol 1992;20:3-16.
- Cooper G. Basic determinants of myocardial hypertrophy: a review of molecular mechanisms. Annu Rev Med 1997;48:13-23.
- Schurch W, Seemayer TA, Gabbiani G. Myofibroblast. In: Sternberg SS, editor. Histology for pathologists. New York: Raven Press; 1992. p 109–44.
- Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin–angiotensin–aldosterone system. Circulation 1991;83:1849-65.
- Fujisaki H, Ito H, Hirata Y. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest 1995;96:1059-65.
- Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res 1998;40:352-63.
- Sano M, Fukuda K, Sato T. ERK and p38 MAPK, but not NF-κB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 2001;89:661-9.
- Dorfman DM, Wilson DB, Bruns GA, Orkin SH. Human transcription factor GATA-2. Evidence for regulation of prepro-endothelin-1 gene expression in endothelial cells. J Biol Chem 1992;267:1279-85.
- Griendling KK, Murphy TJ, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation 1993;87:1816-28.
- Osterrieder W, Muller RK, Powell JS, Clozel JP, Hefti F, Baumgartner HR. Role of angiotensin II in injury-induced neointima formation in rats. Hypertension 1991;18 Suppl II:II-60-II-64.
- Miano J, Topouzis S, Majesky M, Olson E. Retinoid receptor expression and all-trans retinoic acid-mediated growth inhibition in vascular smooth muscle cells. Circulation 1996;93:1886-95.
- Datta PK, Lianos EA. Retinoic acids inhibit inducible nitric oxide synthase expression in mesangial cells. Kidney Int 1999;56:486-93.
- Neuville P, Yan Z, Gidlof A, Pepper MS, Hansson GK, Gabbiani G, et al. Retinoic acid regulates arterial smooth muscle cell proliferation and phenotypic features in vivo and in vitro through an RARa-dependent signaling pathway. Arterioscler Thromb Vasc Biol 1999;19:1430-6.
- Simonson M. Anti-AP-1 activity of all-trans retinoic acid in glomerular mesangial cells. Am J Physiol 1994;267:F805-F815.
- Wang HJ, Zhu YC, Yao T. Effects of all-trans retinoic acid on angiotensin II-induced myocyte hypertrophy. J Appl Physiol 2002;92:2162-8.
- Lü L, Yao T, Zhu YZ, Huang GY, Cao YX, Zhu YC. Chronic all-trans retinoic acid treatment prevents medial thickening of intramyocardial and intrarenal arteries in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2003;285:H1370-H1377.
- Mercola M, Wang CY, Kelly J, Brownlee C, Jackson-Grusby L, Stiles C, et al. Selective expression of PDGF A and its receptor during early mouse embryogenesis. Dev Biol 1990;138:114-22.
- Jakowlew SB, Cubert J, Danielpour D, Sporn MB, Roberts AB. Differential regulation of the expression of transforming growth factor-beta mRNAs by growth factors and retinoic acid in chicken embryo chondrocytes, myocytes, and fibroblasts. J Cell Physiol 1992;150:377-85.
- Glick AB, McCune BK, Abdulkarem N, Flanders KC, Lumadue A, Smith JM, et al. Complex regulation of TGF-β expression by retinoic acid in the vitamin A-deficient rat. Development 1991;111:1081-6.
- Haxsen V, Adam-Stitah S, Ritz E, Wagner J. Retinoids inhibit the actions of angiotensin II on vascular smooth muscle cells. Circ Res 2001;88:637-45.
- Jalil JE, Ebensperger R, Melendez J, Acevedo E, Sapag-Hagar M, Gonzalez-Jara F, et al. Effects of antihypertensive treatment on cardiac IGF-1 during prevention of ventricular hypertrophy in the rat. Life Sci 1999;64:1603-12.
- Pfeffer MA, Pfeffer JM, Froehlich ED. Pumping ability of the hypertrophying left ventricle of the spontaneously hypertensive rat. Circ Res 1976;38:423-9.
- Lijnen PJ, Petrov VV. Role of intracardiac renin-angiotensin-aldosterone system in extracellular matrix remodeling. Methods Find Exp Clin Pharmacol 2003;25:541-64.
- Strickland S, Mahdavi V. The induction of differentiation in terato carcinoma stem cells by retinoic acid. Cell 1978;15:393-403.
- van Giezen JJ, Boon GI, Jansen JW, Bouma BN. Retinoic acid enhances fibrinolytic activity in vivo by enhancing tissue type plasminogen activator (t-PA) activity and inhibits venous thrombosis. Thromb Haemost 1993;69:381-6.
- Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harlev E, et al. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994;372:107-10.
- James TW, Wagner R, White LA, Zwolak RM, Brinckerhoff CE. Induction of collagenase and stromelysin gene expression by mechanical injury in a vascular smooth muscle-derived cell line. J Cell Physiol 1993;157:426-37.
- Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-β. Mol Gen Metab 2000;71:418-35.
- Booz GW, Baker KM. Molecular signaling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res 1995;30:537-43.