
Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats1
Introduction
Pulmonary hypertension (PH) is a common complication of congenital heart defects, with a left-to-right shunt characterized by high pulmonary blood flow. Pulmonary vascular structural remodeling (PVSR) is the pathological basis of PH. To date, the pathophysiological features and mechanisms responsible for PH and PVSR induced by increased pulmonary blood flow have not been fully understood. Some studies demonstrated that high pulmonary blood flow could produce high shear stress on pulmonary artery endothelial cells (EC) and smooth muscle cells (SMC), resulting in a maladjustment of balance in maintaining the structure and function of the vascular wall. Nitric oxide (NO) and carbon monoxide (CO) were found to play an important regulatory role in the development of PH and PVSR[1–3].
Hydrogen sulfide (H2S), a well-known toxic gas with the smell of rotten eggs whose toxicity was first described in 1713, was proven to be generated in vivo in human and animal organisms and participates in many pathophysiological processes. H2S, as a novel gaseous messenger molecule, is generated endogenously in mammalian tissues from L-cysteine metabolism mainly by 3 enzymes: cystathionine β-synthetase (CBS), cystathionine γ-lyase (CSE) and 3-mercaptosulfurtransferase (MST)[4–6]. The distribution of these 3 enzymes is tissue specific, and CSE seems to be the only rate-limiting enzyme to catalyze H2S metabolism in the cardiovascular system[4]. Previous studies by our laboratory and other researchers showed that H2S played a regulatory role in pathophysiological processes in the cardiovascular[7–12], nervous[13] and other systems[14,15]. However, the potential significance of H2S in the pathogenesis of PH and PVSR was unclear. NO is a vasorelaxant factor produced mainly in vascular endothelial cells, while CO and H2S are smooth muscle cell-derived factors. As small gas molecules, they are characterized by common features such as low molecular weight, continuous release and quick dispersal and absorbance, which makes it possible for them to interact. Previous studies, including ours, indicated the possible interaction between NO and H2S[8,16,17] and CO and H2S[18,19] under physiological and pathological conditions. Therefore, the present study was designed to determine whether H2S was involved in the development of PH and PVSR and whether H2S had an impact on the NO/NO synthase (NOS) pathway and CO/heme oxygenase (HO) pathway in a rat model of PH and PVSR induced by high pulmonary flow.
Materials and methods
Animal model of pulmonary hypertension The experiment was conducted in accordance with the Guide for the Care and Use of Experimental Animals issued by the Ministry of Health of the People’s Republic of China. Male Sprague-Dawley rats were provided by the Animal Research Centre of Peking University First Hospital. The rats were housed in plastic cages in a room with a controlled humidity of 40%, at temperature of 22 °C and 12 h light-cycle from 06:00 to 18:00. The animal model was established by an abdominal aorta-inferior vena cava shunt operation according to the method described by Garcia and Diebold[20], with some modifications[21]. Thirty two rats, weighing 120–140 g, were randomly divided into the following groups: shunt (n=8), shunt+NaHS (sodium hydrosulfide) (n=8), sham (n=8) and sham+NaHS (n=8). Rats in the shunt and shunt+NaHS groups were anesthetized with 0.25% pentobarbital sodium (40 mg/kg, intraperitoneal injection). The abdominal aorta and inferior vena cava were exposed, and then a bulldog vascular clamp was placed across the aorta cauda to the left renal artery. The aorta was punctured at the union of the segment two-thirds caudal to the renal artery and one-third cephalic to the aortic bifurcation with use of an 18-gauge disposable needle. The needle was then slowly withdrawn and a 9–0 silk thread was used to stitch the puncture of the abdominal wall. Rats in the sham and sham+NaHS groups underwent the same experimental protocol as mentioned above except for the shunt procedure. Rats in the shunt+NaHS and sham+NaHS groups were intraperitoneally injected with NaHS at 56 μmol/kg each day for 11 weeks as described[7]. Rats in the shunt and sham groups were injected with the same volume of physiological saline.
Blood gas analysis and measurement of Qp/Qs Blood samples (0.5 mL) were obtained from the pulmonary artery, external carotid artery and jugular vein for blood gas analysis with use of a Blood Gas Analysis Apparatus (GASTAT-3, Japan). Qp/Qs was calculated by the formula: Qp/Qs=[oxygen saturation of aorta (%)-oxygen saturation of inferior vena cava (%)]/[oxygen saturation of pulmonary vein (%)-oxygen saturation of pulmonary artery (%)]. When the oxygen saturation of the aorta was >95%, the oxygen saturation of the pulmonary vein was considered 100%. When the oxygen saturation of the aorta was <95%, the oxygen saturation of pulmonary vein was considered 95%. The ratio Qp/Qs was calculated as an indicator to evaluate the pulmonary flow and body flow.
Measurement of hemodynamic parameters and sample preparation At the end of the experiment, the animals were weighed and anesthetized with pentobarbital sodium (40 mg/kg, intraperitoneal injection). A silicone catheter (0.9 mm in outer diameter) was induced under fluoroscope into the right jugular vein via a venotomy and passed across the tricuspid valve and right ventricle into the pulmonary artery. The other end of the catheter was connected to a transducer (YZ-05-1, Beijing, China). The systolic pulmonary artery pressure (SPAP) was simultaneously recorded. Then the heart was removed and the right ventricle (RV) and the left ventricle (LV) plus septum (SP) were dissected. These tissues were weighed on an electronic scale. The ratio of wet weight of right ventricle to left ventricle plus SP [RV/(LV+SP)] was calculated as an indicator of right ventricular hypertrophy. The right side of the lung tissue was removed and kept in liquid nitrogen for quick freezing and then stored at -80 ºC for homogenate. The left lower part of lung tissue was removed to 10% (w/v) paraformaldehyde for fixation. A small part of lung tissue was cut from the upper part of the left lung lobe and quickly immersed into 3% glutaraldehyde.
Morphological analysis of small pulmonary arteries Lung tissue fixed in 10% paraformaldehyde was dehydrated, embedded in paraffin, and cut in sections 4-µm thick. The elastic fiber in lung tissues was stained according to the modified Weigert’s method and counterstained with Van Gieson solution. Morphological analysis was by use of a video-linked microscope digitizing board system (Leica Q550CW, Germany). Only vessels showing clearly defined external and internal elastic lamina were analyzed. In accordance with Bath et al[22], the relative medial thickness (RMT) and relative medial areas (RMA) of intra-acinar pulmonary arteries were calculated. The ratio of muscular artery (MA), partially muscular artery (PMA) and non-muscular artery (NMA) were also observed. Lung tissue fixed in 3% glutaraldehyde was cut into 1 mm×1 mm×1 mm pieces and post-fixed in 1% phosphate-buffered osmium tetroxide for 6 h, then rinsed again (overnight) and dehydrated in a graded series of ethanol. These specimens were infiltrated with propylene oxide and embedded with Epon 812. These procedures were all done at room temperature. Finally, specimens were embedded in new batches of Epon 812 and polymerized at 40 ºC (24 h) and 60 ºC (48 h). Series of transverse or longitudinal sections were obtained from blocks. Semi-thin sections (1 µm) were stained with azur-II and methylene blue. Ultra-thin sections (60–90 nm) were made with use of an ultra microtome and mounted on formvar-coated copper grids (75 meshes), then stained with uranylacetate and lead citrate and examined under a transmission electron microscope (JEM-100CX, JEOL, Japan) in detail.
Western blotting Lung tissues were lysed in a lysis buffer (1 mol/L Tris-Cl 0.2 mL, pH 8.0, 5 mol/L NaCl 0.3 mL, 500 mmol/L edetic acid 10 µL, 100 mmol/L PMSF 0.1 mL, Triton X-100 10 µL, adding water to 10 mL). The extracts were clarified by centrifugation at 12 000 rpm for 15 min at 4 ºC. SDS-PAGE and Western blot analysis was performed according to experimental protocol. The primary antibody dilutions were as follows: 1:200 for eNOS (Santa Cruz Biotechno-logy, Santa Cruz, CA, USA), 1:100 for HO-1 (Sigma, St Louis, MO, USA), 1:200 for extracellular signal-regulated kinase (ERK) (Santa Cruz Biotechnology), 1:200 for phosphorylation ERK (P-ERK) (Santa Cruz Biotechnology), 1:200 for proliferation cell nuclear antigen (PCNA) (Santa Cruz Biotechnology) and 1:5000 for β-actin (TBD, USA). HRP-conjugated secondary antibody was used at 1:5000. The immunoreactions were visualized by ECL and exposed to X-ray film (Kodak Scientific Imaging film, X-omat Blue XB-1, USA).
Assay of H2S in lung tissue Lung tissue was homogenized in a 10-fold volume (w/v) of an ice-cold potassium phosphate buffer (pH 6.8). The reaction was performed in 25 mL in an Erlenmeyer Pyrex flask. Cryovial test tubes (2 mL) were used as the center wells, each containing 1 mol/L NaOH of 0.5 mL. The reaction mixture contained lung tissue homogenate and 1 mol/L HCl at a ratio of 1:5. The flasks containing reaction mixture and central wells were flushed with N2 30 s before being sealed with a double layer of parafilm. The reaction was initiated by transferring the flasks from ice to a shaking water bath at 37 °C. After incubation at 37 °C for 4 h, the contents of the central wells were then transferred to 10-mL beakers, each containing 0.5 mL of antioxidant solution. Subsequently, the solution was measured with sulfide-sensitive electrodes (PXS270, Shanghai, China), and the lung tissue H2S content was calculated against the calibration curve of the standard H2S solution.
Measurement of NO and NOS activity in lung tissue In accordance with the methods described by Stathopulos et al[23], with modification, NO production and NOS activity in lung tissue were measured at 540 nm with use of a Spectrophotometer (DU 6400; Beckman).
Measurement of CO in lung tissue CO content in lung tissue was detected according to the methods described by Morita and Kourembanas[24] and Chalmer[25], with modifica-tion. The lung tissue homogenate was centrifuged at 12 000 rpm for 20 min and 0.5 mL of the supernatant was mixed with 1 mL of the hemoglobin solution, which was derived from a mixture of 0.25 mL of fresh packed erythrocytes from rats and 50 mL of 0.25 mol/L ammonia solution. Then 0.1 mL of sodium dithionite was added. The absorbance of the tested lung tissue homogenate sample and water blank at 568 nm and 581 nm was read and the ratio (R) of absorbance at the 568 and 581 nm readings was calculated. Then the percentage of HbCO from a standard curve derived by mixing different proportions of 2 Hb solutions containing 100% HbCO and 100% HbO2 was derived. Lung tissue homogenate CO was then calculated as follows: CO (nmol/µg)=% HbCO×Hb (mg/L)×4×4×1000/(64 456×100×X). X represents the volume of homogenate.
Statistical analysis Data were expressed as mean±SD and statistically analyzed by use of SPSS 10.0. Comparison among groups involved one-way ANOVA followed by Student-Neuman-Keuls’ tests. A value of P<0.05 was considered statistically significant.
Results
Changes in Qp/Qs and hemodynamic parameters In the present rat model of abdominal aorta-inferior vena cava shunt, Qp/Qs in the shunt-alone group and shunt+NaHS group increased significantly as compared with that of sham group (P<0.01). The shunt and shunt+NaHS groups did not differ significantly in Qp/Qs. As shown in Table 1, the SPAP increased significantly in rats of the shunt-alone group as compared with that of the sham group (P<0.01), whereas the SPAP in rats of the shunt+NaHS group decreased significantly as compared with that of the shunt-alone group (P<0.01). The SPAP between the sham and sham+NaHS groups, however, did not differ significantly. RV/(LV+SP) was higher in rats of shunt-alone group than that of the sham group (P<0.01). Whereas
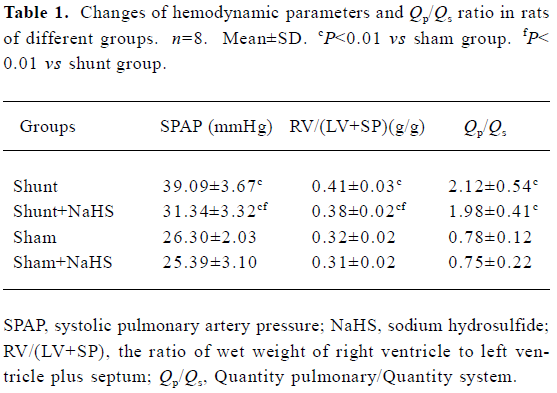
Full table
Administration of exogenous H2S donor alleviated pulmonary vascular structural remodeling As shown in Table 2, the proportion of MA and PMA increased significantly, whereas NMA decreased significantly in the rats of the shunt-alone group as compared with that of sham group (all P<0.01). After the administration of exogenous H2S donor for 11 weeks, the proportion of MA and PMA decreased significantly, and that of NMA increased as compared with the shunt-alone group (all P<0.01). The sham and sham+ NaHS groups did not differ significantly in proportion to MA, PMA, and NMA.
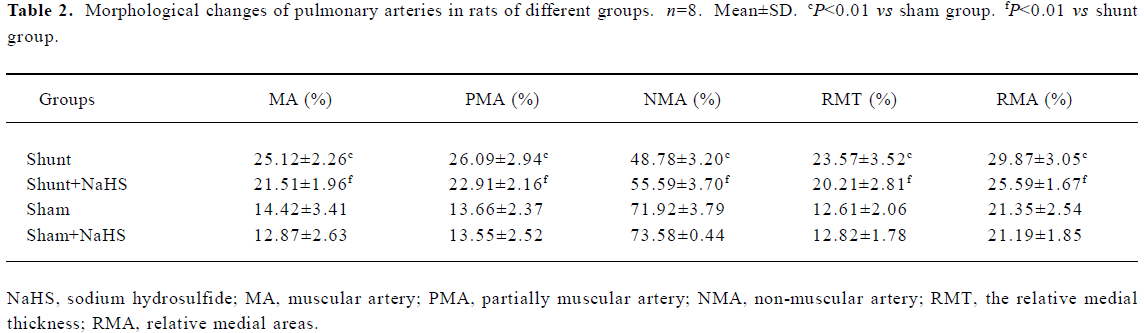
Full table
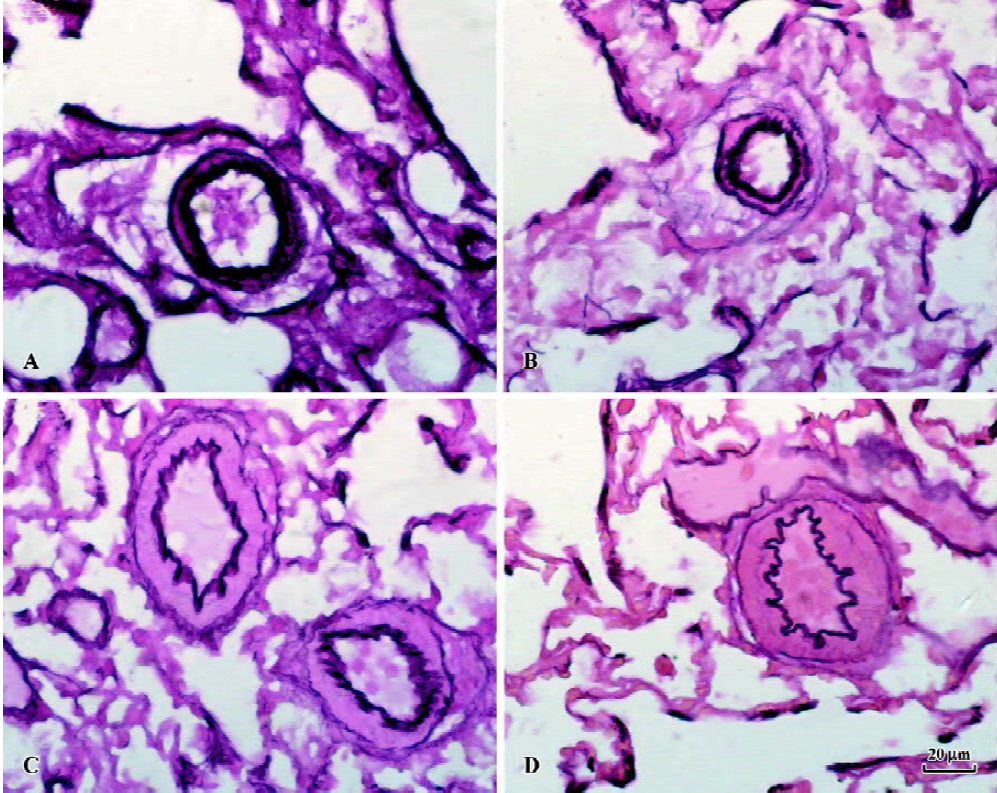
RMT and RMA of intra-acinar pulmonary arteries increased significantly in rats of shunt-alone group as compared with that of the sham group (all P<0.01). After administration of NaHS for 11 weeks, RMT and RMA decreased significantly as compared with shunt-alone group (all P<0.01). The sham and sham+NaHS groups did not differ significantly in RMT and RMA proportions (Figure 1).
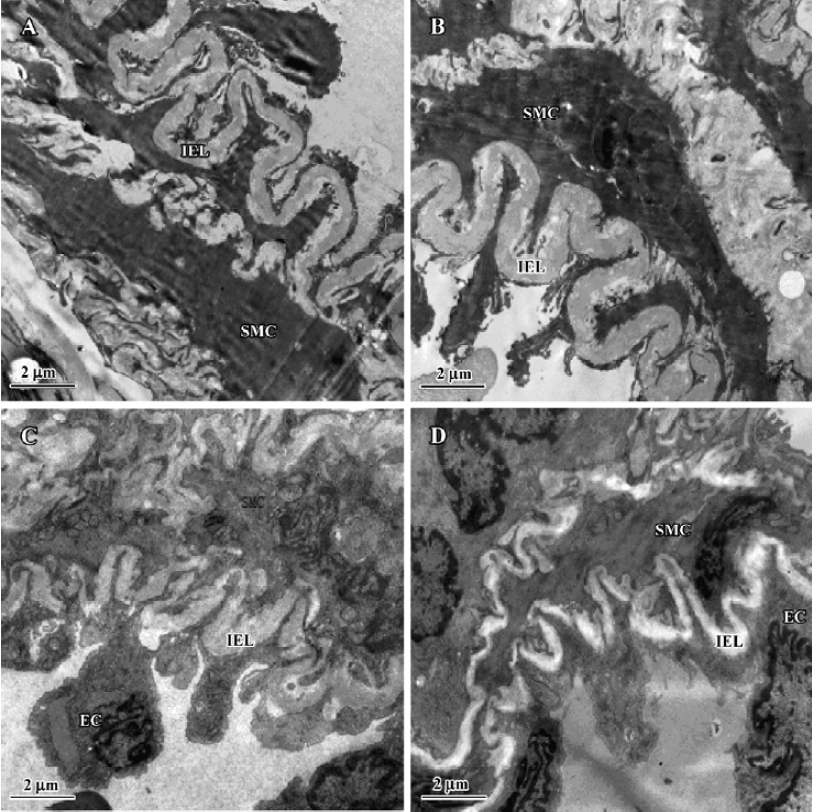
After 11 weeks of shunting, the ultra-structure of the intraacinar pulmonary arteries showed swollen and hypertrophic endothelial cells, irregular internal elastic lamina and increased rough endoplasmic reticulum and free ribosomes in the cytoplasm of SMC. After administration of NaHS for 11 weeks, the intraacinar pulmonary arteries showed the flat endothelial cells, regular internal elastic lamina and cytoplasm of SMC full of myofilaments and dense bodies (Figure 2).
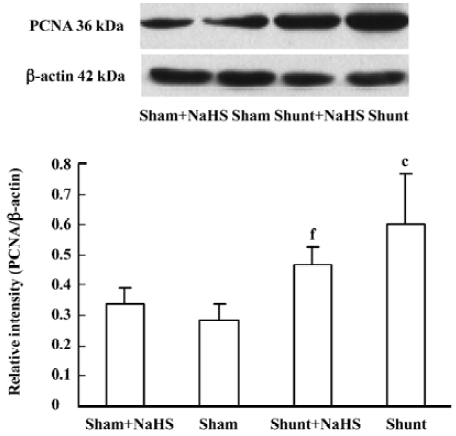
Administration of exogenous H2S donor impacted pulmonary vascular structural remodeling and ERK/MAPK pathway PCNA expression of the pulmonary artery increased significantly in rats of the shunt-alone group as compared with that of the sham group (P<0.01). After administration of NaHS for 11 weeks, compared with that of the shunt-alone group, the shunt+NaHS group showed significantly decreased PCNA expression in pulmonary artery (P<0.01). The sham and sham+NaHS groups did not differ significantly in PCNA expression of the pulmonary artery (Figure 3).
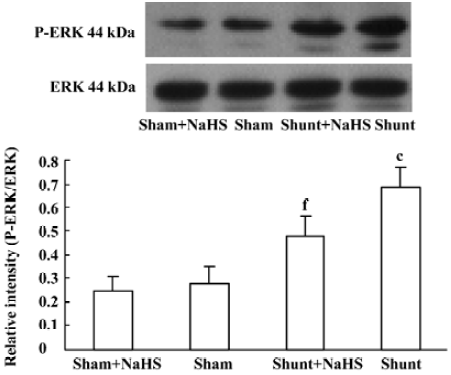
The ratio of P-ERK/ERK1 expression of pulmonary arteries increased significantly in rats of the shunt-alone group as compared with that of the sham group (P<0.01). After administration of NaHS for 11 weeks, the ratio of P-ERK/ERK1 expression of pulmonary arteries decreased significantly as compared with the shunt-alone group (P<0.01). The sham and sham+NaHS groups did not differ significantly in the ratio of P-ERK/ERK1 expression of the pulmonary artery (Figure 4).
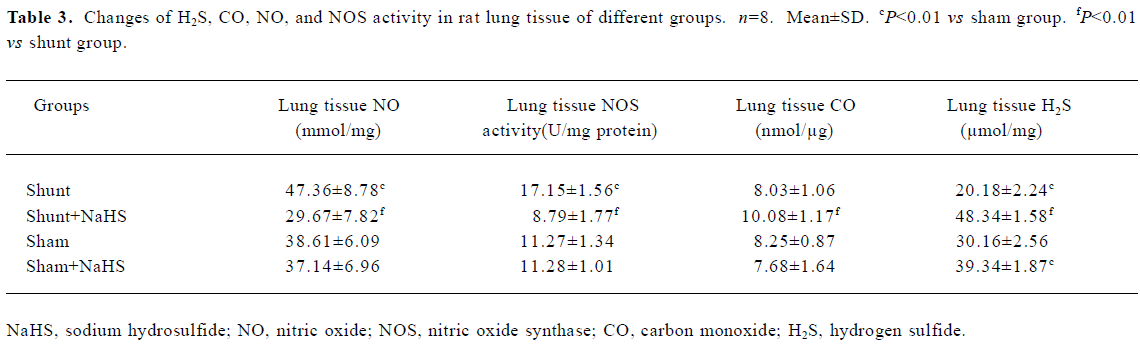
Administration of exogenous H2S donor increased the level of lung tissue H2S The production of lung tissue H2S in rats of the shunt-alone group was lower than that of the sham group. After administration of NaHS for 11 weeks in rats of the shunt+NaHS and sham+NaHS groups, the level of H2S increased significantly (P<0.01) (Table 3).
Full table
Administration of exogenous H2S donor attenuated the increased production of endogenous NO, NOS activity and eNOS protein in lung tissue As shown in Table 3, the production of endogenous NO increased significantly in rats of the shunt-alone group as compared with those of the sham group (P<0.01). After treatment with NaHS for 11 weeks, the production of endogenous NO decreased significantly compared with the shunt-alone group (P<0.01). The sham and sham+NaHS groups did not differ significantly in the production of endogenous NO. Corresponding to the changes in the production of endogenous lung tissue NO, NOS activity increased significantly in rats of the shunt-alone group as compared with that of the sham group (P<0.01). In contrast to the shunt-alone group, the shunt+NaHS group showed that NOS activity decreased by 95.11% in lung tissue (P<0.01). The sham and sham+NaHS groups did not differ significantly in NOS activity in lung tissue (Table 3).
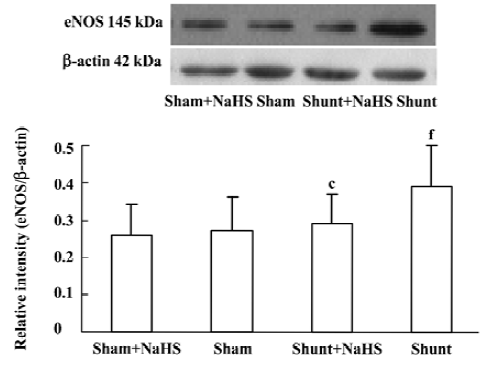
Lung tissue eNOS protein expression in rats of the shunt-alone group was up-regulated significantly compared with that of the sham group (P<0.01). In contrast to the shunt-alone group, the shunt+NaHS group showed significantly down-regulated eNOS protein expression in lung tissue (P<0.01). The sham and sham+NaHS groups did not differ significantly in eNOS protein expression in lung tissue (Figure 5).
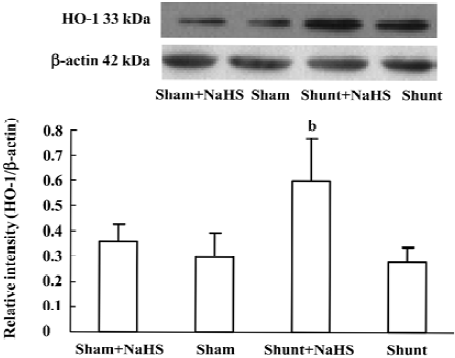
Administration of exogenous H2S donor increased the production of endogenous CO and expression of HO-1 protein in lung tissue The production of endogenous CO between the sham and shunt-alone groups did not significantly differ. In contrast to the shunt-alone group, the shunt+NaHS group showed a significant increase in the production of endogenous CO (P<0.01). The sham and sham+NaHS groups did not differ significantly in the production of endogenous CO (Table 3).
The changes in lung tissue HO-1 protein expression were consistent with those of the production of endogenous CO. The lung tissue HO-1 protein expression between the sham and shunt-alone groups did not differ significantly. Lung tissue HO-1 protein expression increased significantly in rats of the shunt+NaHS group as compared with that of the shunt-alone group (P<0.01). The sham and sham+NaHS groups did not differ significantly in the lung tissue HO-1 protein expression (Figure 6).
Discussion
In the present study, we successfully established the rat model of aortocaval shunt. The mortality was 5.9% of the 17 rats with an aortocaval shunt one died within 24 h of shunt implantation because of a congested lung as was seen on autopsy.
After 11 weeks of shunting, the SPAP increased by 48.63%, which suggested that shunt rats with high pulmonary blood flow developed PH. Furthermore, the proportion of MA and PMA increased by 74.20% and 90.99%, respectively, whereas that of NMA decreased by 32.17%, and the RMT and RMA of the intra-acinar pulmonary arteries increased by 86.92% and 39.91%, respectively. The ultrastructure of the intra-acinar pulmonary arteries showed swollen and hypertrophic endothelial cells, irregular internal elastic lamina and increased rough endoplasmic reticulum and free ribosomes in the cytoplasm of SMC. The above findings demonstrated that PVSR was characterized by increased mascularization and ultrastructural changes of small pulmonary arteries, along with the development of PH.
The mechanisms responsible for PH and PVSR induced by high pulmonary flow have not been fully understood. Studies showed that endothelial cells of pulmonary arteries were constantly exposed to fluid mechanical forces derived by high-flowing blood. These forces, especially shear stress, caused progression from endothelial dysfunction to smooth muscle dysfunction as vascular changes progress, resulting in the PH and PVSR[26]. Many cytokines and growth factors, such as basic fibroblast growth factor, transforming growth factor β, epidermal growth factor, platelet derived growth factor and endothelin, are implicated as contributing fact-ors[26–28]. However, the mechanism for PVSR has not been understood. In the 1980s, NO and CO were determined to be gaseous messenger molecules sharing common features such as low molecular weight, continuous release and quick dispersal and absorbance. The discovery that NO has powerful vasoactive properties identical to those of endothelial-derived relaxing factor spawned a vast body of research investigating the physiological actions of small gas molecules in PH and PVSR[29]. Like NO, CO acts as a vasorelaxant and regulates other vascular functions such as smooth muscle proliferation. CO, when applied at low concentrations, exerts potent cytoprotective effects mimicking those of HO-1 induction in PH and PVSR[30]. However, many questions are still unanswered regarding the mechanisms responsible for the development of PH and PVSR.
H2S, the newly discovered third endogenous gasotransmitter, was known as a toxic gas for hundreds of years, with little attention paid to its physiological and pathophysiological function. Studies have demonstrated the biological functions of H2S such as hyperpolarization of cell membranes[4], relaxation of SMC[4], inhibition of SMC proliferation[31], and decrease in neuronal excitability[32]. Our recent studies showed that H2S in some cardiovascular diseases played a regulatory role, such as myocardial injury induced by isoproterenol[12], hypertension induced by inhibition of NOS[8], spontaneous hypertension[7] and hypoxic pulmonary hypertension[9]. However, whether and how H2S plays a role in PH and PVSR induced by high pulmonary blood flow is still unknown.
To seek the answer to this question, we measured the production of lung tissue H2S in shunted rats. After 11 weeks of shunting, PH, and PVSR resulted from high pulmonary blood flow. Interestingly, the production of lung tissue H2S in shunted rats was decreased by 33.09%.
To further clarify the possible role of H2S in PH and PVSR induced by high pulmonary blood flow, NaHS, a donor of exogenous H2S was supplied for 11 weeks, and it significantly increased the production of lung tissue H2S. At the same time, SPAP decreased by 19.82% and RV/(LV+SP) decreased by 7.3%, which suggested that the increased H2S could decrease pulmonary pressure and attenuate right ventricular hypertrophy in rats with high pulmonary flow. Also, after administration of NaHS for 11 weeks, the proportion of MA and PMA decreased by 14.37% and 12.18%, respectively, and that of NMA increased by 13.96%. RMT and RMA decreased by 14.26% and 14.33%, respectively. The intra-acinar pulmonary arteries showed flat endothelial cells, regular internal elastic lamina and a cytoplasm of SMC full of myofilaments and dense bodies. The findings above demonstrated that NaHS alleviated PVSR induced by high pulmonary blood flow in rats.
However, the mechanisms by which H2S regulates PH and PVSR are not fully understood. Vascular smooth muscle cell (VSMC) proliferation resulted in vascular wall thickening and increased pulmonary vascular resistance, which is the pathophysiological base of PH and PVSR. Our previous in vitro study showed that H2S could inhibit VSMC proliferation in the cultured cells of rats[27]. Yang et al reported that CSE overexpression could inhibit cell proliferation in vitro[33]. However, whether H2S affects VSMC proliferation and its roles in vivo have not been reported. In this experiment, we found that PVSR was characterized by thickened medial membrane in shunted rats, and administration of NaHS for 11 weeks alleviated the thickening, as shown by thinned medial membrane in shunted rats. To explore whether PVSR is involved in VSMC proliferation, PCNA expression was investigated. After 11 weeks of shunting, PCNA protein expression of the pulmonary artery increased significantly. After administration of NaHS for 11 weeks, PCNA protein expression decreased significantly in pulmonary arteries. The results suggest that exogenous H2S attenuated the increased pulmonary SMC proliferation in rats with high pulmonary blood flow in vivo.
The MAPK cascade plays a crucial role in the transduction of extracellular signals into responses governing growth and differentiation. MAPK-catalyzed phosphorylation of substrate proteins functions as a switch to turn on or off the activity of the substrate protein. The ERK pathway has been shown to be involved in the stimulation of cellular proliferation[34]. The ratio of P-ERK/ERK indicates the degree of activation of the ERK/MAPK pathway. In this experiment, after 11 weeks of shunting, the ratio of P-ERK/ERK1 in pulmonary arteries increased significantly. After the administration of NaHS for 11 weeks, the ratio of P-ERK/ERK1 in pulmonary arteries decreased significantly. Nevertheless, whether H2S inhibited VSMC proliferation by the ERK/MAPK pathway in rats with high pulmonary blood flow in vivo needs to be studied further.
NO and CO are both important gasotransmitters formed in vessels. They play a part in the regulation of PVSR. Our previous study showed a possible interaction between H2S and NO and between H2S and CO in blood vessels[17,18,35]. However, little is known about the impact of H2S on the NO/NOS and CO/HO pathways in a rat model of PH and PVSR induced by high pulmonary blood flow. To further understand the possible mechanism by which H2S regulated PH and PVSR in rats with high pulmonary blood flow, we investigated the lung tissue NO, NOS activity and eNOS protein. It was shown that, after 11 weeks of shunting, the production of endogenous NO, NOS activity and eNOS protein expression increased significantly. After treatment with NaHS for 11 weeks, the production of endogenous NO, NOS activity and eNOS protein expression decreased significantly. The above findings showed that exogenous H2S attenuated the activated NO/NOS pathway in shunted rats, which might be involved in the mechanism by which H2S alleviates PVSR.
CO, another gaseous small molecule, has been reported to inhibit VSMC proliferation in hypoxic pulmonary hypertension[3]. In the present rat model, we found that, after treatment with NaHS for 11 weeks, the production of endogenous CO increased significantly and HO-1 protein expression in lung tissue was up-regulated significantly.
How H2S affects the NO/NOS and CO/HO pathways is still unknown in the pathogenesis of PH induced by high pulmonary blood flow. NO, CO, and H2S were considered as gasotransmitters, therefore we postulated that NO, CO, and H2S might possibly interact under physiological and pathological conditions, and they might constitute a regulatory network in the vascular system. In the present study, NaHS increased the level of H2S but decreased the generation of lung tissue NO, NOS activity and eNOS protein expression, which suggested that H2S might inhibit the generation of endogenous NO through down-regulation of eNOS protein expression and decreased NOS activity. However, whether changes in the NO pathway are involved in NaHS regulating PVSR needs to be studied further.
NaHS increased the level of lung tissue H2S but facilitated the generation of CO, by up-regulating HO-1 protein expression. H2S can be scavenged by methemoglobin, or metallo- or disulfide-containing molecules such as oxidized glutathione[36]. Hemoglobin may be the common “sink” for CO in forming carboxyhemoglobin[37], and for H2S in forming green sulhemoglobin. We speculate that H2S might directly affect carboxyhemoglobin metabolism resulting in the release of CO under pathological conditions. Nevertheless, the molecular mechanism by which H2S regulates the CO/HO pathway needs to be studied further.
H2S also has a direct effect on the vasculature, which might be involved in the mechanisms by which H2S attenuated PH. Previous studies showed that H2S exerted a direct effect on vessels as a vasodilator in vitro[4,16], and its mechanism was related to the opening of the KATP channel .
NaHS was used as a donor of H2S in this study for the following reasons: (1) It was reported that about one-third of H2S existed as the undissociated form (H2S) and the remaining two-thirds as HS– at equilibrium with H2S in physiological saline[27]. NaHS dissociates to Na+ and HS- in solution and then HS- associates with H+ and produces H2S. (2) A bolus intraperitoneal injection of NaHS leading to an increase in plasma H2S level and a decrease in mean arterial blood pressure continued at least for 6 h in rats[8], much longer than a transient decrease in blood pressure (lasting minutes) caused by an intravenous bolus injection of gaseous H2S [4], and the use of NaHS enables us to define the concentrations of H2S in solution more accurately and reproducibly than bubbling H2S gas. (3) NaHS at concentrations used in the present study does not change the pH of the medium.
In conclusion, this study provided evidence for the first time that H2S exerted a regulatory effect on PH and PVSR induced by high pulmonary blood flow. Meanwhile, H2S down-regulated the ERK/MAPK signal pathway, inhibited the NO/NOS pathway and enhanced the CO/HO pathway in rats with high pulmonary blood flow. Whether H2S could be a therapeutic target for PH and PVSR induced by high pulmonary blood flow needs further exploration.
References
- Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, et al. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res 2000;86:1224-9.
- Wei B, Du JB, Qi JG, Li J, Tang CS. L-Arginine impacts pulmonary vascular structure in rats with an aortocaval shunt. J Surg Res 2002;108:20-31.
- Shi Y, Du JB, Gong LM, Zeng CM, Tang XY, Tang CS. The regulating effect of heme oxygenase/carbon monoxide on hypoxic pulmonary vascular structural remodeling. Biochem Biophys Res Commun 2003;306:523-9.
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 2001;20:6008-16.
- Nagahara N, Ito T, Kitamura H, Nishino T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem Cell Biol 1998;110:243-50.
- Ogasawara Y, Isoda S, Tanabe S. Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biol Pharm Bull 1994;17:1535-42.
- Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 2004;13:22-7.
- Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 2003;21:1879-85.
- Zhang CY, Du JB, Bu DF, Yan H, Tang XY, Tang CS. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun 2003;302:810-6.
- Hui Y, Du J, Tang C, Bin G, Jiang H. Changes in arterial hydrogen sulfide (H2S) content during septic shock and endotoxin shock in rats. J Infect 2003;47:155-60.
- Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J, et al. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 2004;313:362-8.
- Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, et al. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun 2004;318:756-63.
- Kimura H. Hydrogen sulfide as a neuromodulator. Mol Neurobiol 2002;26:13-9.
- Fiorucci S, Antonelli E, Mencarelli A, Orlandi S, Renga B, Rizzo G, et al. The third gas: H(2)S regulates perfusion pressure in both the isolated and perfused normal rat liver and in cirrhosis. Hepatology 2005;42:539-48S.
- Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J 2005;19:1196-8.
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 1997;237:527-31.
- Zhang QY, Du JB, Shi L, Zhang CY, Yan H, Tang CS. Interaction between endogenous nitric oxide and hydrogen sulfide in pathogenesis of hypoxic pulmonary hypertension. Beijing Da Xue Xue Bao 2004;36:52-6.
- Zhang QY, Du JB, Zhou WJ, Yan H, Tang CS, Zhang CY, et al. Impact of hydrogen sulfide on carbon monoxide oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 2004;317:30-7.
- Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol 2006;100:1065-76.
- Garcia R, Diebold S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res 1990;24:430.
- Ocampo C, Ingram P, Ilbawi M, Arcilla R, Gupta M. Revisiting the surgical creation of volume load by aorto-caval shunt in rats. Mol Cell Biochem 2003;251:139-43.
- Bath PJ, Kimpel CH, Roy S, Wagner U. An improved mathematical approach for the assessment of the medical thickness of pulmonary arteries. Pathol Res Pract 1993;189:567-76.
- Stathopulos PB, Lu X, Shen J, Scott JA, Hammond JR, McCormack DG, et al. Increased l-arginine uptake and inducible nitric oxide synthase activity in aortas of rats with heart failure. Am J Physiol 2001;280:H859-67.
- Morita T, Kourembanas S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest 1995;96:2676-82.
- Chalmers AH. Simple, sensitive measurement of carbon monoxide in plasma. Clin Chem 1991;37:1443-5.
- Bongrazio M, Baumann C, Zakrzewicz A, Pries AR, Gaehtgens P. Evidence for modulation of genes involved in vascular adaptation by prolonged exposure of endothelial cells to shear stress. Cardiovasc Res 2000;47:384-93.
- Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular matrix-bound basic fibroblast growth factor. J Cell Physiol 1995;166:495-505.
- Dardik A, Yamashita A, Aziz F, Asada H, Sumpio BE. Shear stress-stimulated endothelial cells induce smooth muscle cell chemotaxis via platelet-derived growth factor-BB and interleukin-1alpha. J Vasc Surg 2005;41:321-31.
- Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 2006. [Epub ahead of print].
- Hartsfield CL, McMurtry IF, Ivy DD, Morris KG, Vidmar S, Rodman DM, et al. Cardioprotective and vasomotor effects of HO activity during acute and chronic hypoxia. Am J Physiol Heart Circ Physiol 2004;287:H2009-15.
- Du J, Hui Y, Cheung Y, Bin G, Jiang H, Chen X, et al. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels 2004;19:75-80.
- Eto K, Ogasawara M, Umemura K, Nagai Y, Kimura H. Hydrogen sulfide is produced in response to neuronal excitation. J Neuro Sci 2002;22:3386-91. a.
- Yang G, Cao K, Wu L, Wang R. Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem 2004;279:49199-205.
- Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA 1993;90:5889-92.
- Shi Y, Du JB, Guo ZL, Zeng CM, Tang CS. Interaction between endogenous nitric oxide and carbon monoxide in the pathogenesis of hypoxic pulmonary hypertension. Chin Sci Bull 2003;48:86-90.
- Beauchamp RO, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol 1984;13:25-97.
- Wang R. Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor. Can J Physiol Pharmacol 1998;76:1-15.