
Simvastatin inhibits leptin-induced hypertrophy in cultured neonatal rat cardiomyocytes1
Introduction
Obesity is a major risk factor for the development of cardiovascular diseases such as hypertension, atherosclerosis, and heart failure[1,2]. Leptin is the product of the ob gene, and its concentration is increased in obese individuals. Animal studies indicate that long-term administration of leptin results in a sustained increase in artery pressure. Clinical evidence indicates that leptin is a potential independent risk factor for coronary heart disease. Recent studies have supplied evidence that leptin also induces hypertrophy in rat neonatal cardiomyocytes in vitro[3,4]. Clinical evidence also indicates that increased plasma leptin levels are correlated with cardiac hypertrophy and congestive heart failure[5,6]. These results suggest that leptin is an important factor in the cause of obesity-related cardiovascular diseases.
Statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), the rate-limiting enzyme of cholesterol synthesis. Emerging evidence from both animal and human studies indicates that mechanisms independent of cholesterol lowering effects contribute to the observed clinical benefits of statins[7,8]. Recent studies (including our own) have demonstrated that statins prevent cardiac hypertrophy and heart failure in animals with aortic stenosis and angiotensin II-infusion[9,10]. However, it is unknown whether statins can inhibit obesity-related cardiac hypertrophy.
In the present study, we hypothesized that simvastatin inhibited leptin-induced cardiomyocyte hypertrophy. Our results suggest that leptin stimulates hypertrophy via reactive oxygen species (ROS) generation in cultured neonatal rat cardiomyocytes. We found that treatment with simvastatin or the antioxidants butylated hydroxyanisole (BHA) and catalase inhibited leptin-induced ROS production and hypertrophy in cultured neonatal rat cardiomyocytes.
Materials and methods
Cell culture Primary cultures of cardiomyocytes were prepared according to previously published methods[11,12]. Briefly, hearts from 1-day-old Sprague-Dawley rats that were obtained from the Medical Laboratory Animal Center (Guangdong, China) were dissected, minced and placed in a Petri dish. The tissue was trypsinized at 37 °C in D-Hanks’ balanced salt solution (in g/L: NaCl 8.00, KCl 0.4, KH2PO4 0.06, NaHCO3 0.35, Na2HPO4.7H2O 0.09 and 0.125% trypsin). After centrifugation (200 g, 15 min), cells were collected and suspended in M199 medium (containing 10% fetal calf serum, insulin 5 µg/mL, transferrin 5 µg/mL, penicillin 100 U/mL, and streptomycin 100 µg/mL (Sigma, St Louis, USA). Cells were then preplated at 37 °C for 1 h. The cells were diluted to 5×106 cells/mL, plated in 96-well, 24-well or 6-well plates, and were cultured for 48–64 h in medium containing 0.1 mmol/L bromodeoxyuridine to prevent proliferation of nonmyocytes. On the fifth day, after being cultured in a non-serum medium for 72 h, cells were washed twice with Hanks’ balanced salt solution at 37 °C. The washed cells were incubated at 37 °C for 4 h and in a non-serum medium containing leptin (100 ng/mL, R&D Systems, Minneapolis, MN, USA) for 72 h, leptin (100 ng/mL) plus simvastatin (1–100 nmol/L, Merck, Sharp & Dohme, NJ, USA), leptin (100 ng/mL) plus the antioxidant butylated hydroxyanisole (BHA; 10 µmol/L; Sigma)[13] or leptin (100 ng/mL) plus catalase (200 U/mL, Sigma), an enzyme that specifically decomposes hydrogen peroxide (H2O2) to water and molecular oxygen.
ROS determination Intracellular ROS was assessed by using the ROS-specific probe 2',7'-dichlorofluorescein diacetate (DCF-DA, Molecular Probes, Eugene, OR, USA)[11,14]. On culture d 4, the cultured cardiomyocytes were washed with Hanks’ solution, and then incubated with DCF-DA (5 mmol/L) in Hanks’ solution at 37 °C. After incubation for 1 h, cardiomyocytes were washed with Hanks’ solution. Fluorescence signals were obtained with a fluorescence microscope (TE300-ECI; Nikon, Tokyo, Japan) and assayed by using its image processing and analysis system. In each case, 5 randomly selected fields in each well were selected for examination.
RNA content measurement The RNA content was determined by using the RNA-sensitive fluorescence probe propidium iodide (PI; Molecular Probes) after DNase treatment[11,15]. Cardiomyocytes were treated with diluent (control), leptin (100 ng/mL), leptin (100 ng/mL) plus simvastatin (1−100 nmol/L), or leptin (100 ng/mL) plus catalase (200 U/mL) from culture d 4 to d 7. On culture d 7, the cells were washed with Hanks’ solution and fixed with 75% ethanol for 10 min. Ethanol-fixed cells were rinsed in Hanks’ solution and incubated at 36 °C for 40 min in a solution containing DNase (1 mg/mL), sucrose (0.25 mol/L), MgCl2 (5 mmol/L), and Tris-HCl (20 mmol/L; pH 6.5). After incubation, 1 mL of Hanks’ solution containing PI (0.05 mg/mL) was added to each well and left for 30 min. Then the fluorescence signal was analyzed with a fluorescence microscope. In each case, five randomly selected fields in each well were selected for examination.
Cardiomyocyte surface area assay Cardiomyocyte surface area was measured according to a previously described method[16]. Cell images were captured with a digital camera (Nikon) fixed to a microscope (Nikon). Cardiomyocyte surface area was analyzed using the Leica Image Processing and Analysis System. One hundred cells from randomly selected fields in 3 wells were examined for each condition. Cardiomyocyte surface area was determined after 3-d treatment with leptin, simvastatin, or the antioxidant catalase in comparison with control cells treated with diluent.
Protein content evaluation Cultured cardiomyocytes were treated with leptin, simvastatin, catalase, or diluent (control) from culture d 4 to d 7. The cells were washed with PBS and then treated with 5% trichloroacetic acid at 4 °C for 1 h to precipitate the protein[11,16]. The precipitates were dissolved in NaOH (0.1 mol/L). The protein content was measured using the Bio-Rad (Hercules, CA, USA) DC protein assay.
Statistical analysis All results are expressed as mean±SD. One-way ANOVA was used for multiple comparisons. A value of P<0.05 was considered significant.
Results
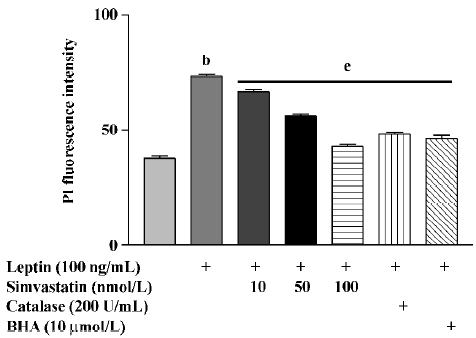
After 72 h treatment, leptin (100 ng/mL) significantly increased RNA levels in cultured cardiomyocytes. Treatment with simvastatin, BHA, or catalase significantly inhibited the effect of leptin on RNA levels (Figure 1).
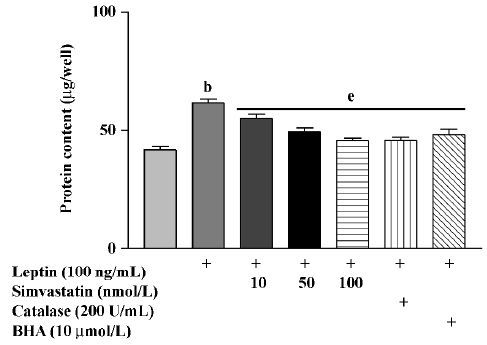
Protein content in cultured cardiomyocytes treated with leptin (100 ng/mL) for 72 h was significantly increased. Treatment with simvastatin, BHA, or catalase markedly inhibited the effect of leptin on protein content (Figure 2).
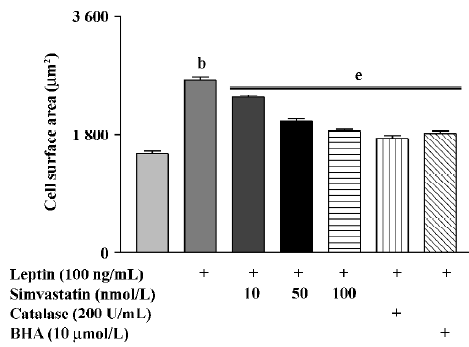
Cell surface area in cultured cardiomyocytes treated with leptin for 72 h was significantly increased. Treatment with simvastatin, BHA, or catalase markedly inhibited the effect of leptin on cell surface area (Figure 3).
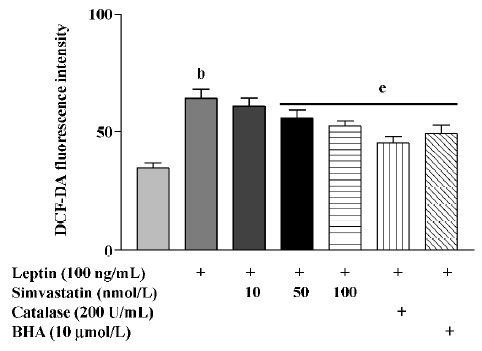
After treatment for 4 h, leptin markedly increased the ROS levels of cultured cardiomyocytes, an effect that was blunted by treatment with simvastatin, BHA, or catalase (Figure 4).
Discussion
The present study demonstrated that leptin induced hypertrophy through a ROS-dependent pathway and that simvastatin inhibited leptin-induced hypertrophy in cultured neonatal rat cardiomyocytes.
Clinical evidence has shown that plasma leptin levels are increased in obese individuals, and that enhanced plasma leptin levels are correlated with cardiac hypertrophy and other cardiovascular diseases[6,17]. This observation is supported by recent studies in vitro[3]. The results of these studies indicate that leptin induces hypertrophy in cultured rat neonatal cardiomyocytes in a concentration-dependent manner. These results provide a link between leptin and cardiac hypertrophy in obese individuals. However, the mechanisms by which leptin induces cardiac hypertrophy are still unknown. Therefore, in the present study, we examined how leptin induced cardiac hypertrophy. Our results indicate that leptin induces ROS generation and hypertrophy, which were significantly inhibited by simvastatin and catalase (an enzyme that specifically decomposes hydrogen peroxide to water and molecular oxygen) in cultured neonatal rat cardiomyocytes. Collectively, these data suggest that oxidative stress is involved in leptin-induced hypertrophy in cultured neonatal rat cardiomyocytes.
It is also well known that oxidative stress contributes to cardiac hypertrophy, because hypertrophic substances such as endothelin-1, norepinephrine, angiotensin II, and cytokines such as tumor necrosis factor (TNF)-α induce cardiomyocyte hypertrophy via ROS generation[11,16]. Furthermore, clinical and animal studies have demonstrated that the failing heart is subjected to increased oxidative stress, and antioxidant therapy has been shown to preserve left ventricular function during the development of chronic heart failure[19,20]. Recent evidence has also indicated that obesity was a state of chronic oxidative stress and inflammation[2,18]. However, it is unknown whether oxidative stress contributes to cardiac hypertrophy in the obese state. Our present results show that leptin induces ROS generation and hypertrophy, which can be reversed by treatment with the antioxidants catalase and simvastatin, in cultured neonatal rat cardiomyocytes. Thus, these results suggest that elevated leptin levels in obese subjects may increase cardiac oxidative stress, which results in cardiac hypertrophy. Therefore, antioxidant therapy aimed at reducing oxidative stress induced by leptin may provide an effective means to improve cardiac dysfunction in obese humans.
Animal studies have also shown that statins induce the regression of cardiac hypertrophy in vivo[9,10] and inhibit the hypertrophy of cardiomyocytes induced by norepinephrine and angiotensin II in vitro[11,16]. Inhibition of the Rac1-ROS pathway represents an important mechanism by which statins inhibit cardiac hypertrophy[21,22]. Our present results indicate that simvastatin inhibits leptin-induced ROS-mediated hypertrophy in cultured neonatal rat cardiomyocytes. The results further show that statins are effective against cardiac hypertrophy in different animal models, and highlight the need for clinical trials to determine the effects of statins on cardiac hypertrophy and fibrosis in humans.
References
- Williams CL, Hayman LL, Daniels SR, Robinson TN, Steinberger J, Paridon S, et al. Cardiovascular health in childhood: a statement for health professionals from the Committee on Athero-sclerosis, Hypertension, and Obesity in the Young (AHOY) of the Council on Cardiovascular Disease in the Young, American Heart Association. Circulation 2002;106:143-60.
- Keaney JF Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterio-scler Thromb Vasc Biol 2003;23:434-9.
- Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res 2003;93:277-9.
- Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, et al. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation 2004;110:1269-75.
- Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, et al. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 2001;104:3052-6.
- Paolisso G, Tagliamonte MR, Galderisi M, Zito GA, Petrocelli A, Carella C, et al. Plasma leptin level is associated with myocardial wall thickness in hypertensive insulin-resistant men. Hypertension 1999;34:1047-52.
- Luo JD, Chen AF. Perspectives on the cardioprotective effects of statins. Curr Med Chem 2003;10:1593-601.
- Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest 2002;110:285-8.
- Luo JD, Zhang WW, Zhang GP, Guan JX, Chen X. Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clin Exp Pharmacol Physiol 1999;26:903-8.
- Dechend R, Fiebeler A, Park JK, Muller DN, Theuer J, Mervaala E, et al. Amelioration of angiotensin II-induced cardiac injury by a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Circulation 2001;104:576-81.
- Luo JD, Xie F, Zhang WW, Ma XD, Guan JX, Chen X. Simvastatin inhibits noradrenaline-induced hypertrophy of cultured neonatal rat cardiomyocytes. Br J Pharmacol 2001;132:159-64.
- Simpson P, McGrath A, Savion S. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ Res 1982;51:787-801.
- Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-α and angiotensin II. Circulation 1998;98:794-9.
- Zhu H, Bannenberg GL, Moldeus P, Shertzer HG. Oxidation pathways for the intracellular probe 2',7'-dichlorofluorescein. Arch Toxicol 1994;68:582-7.
- Frankfurt OS. Flow cytometric analysis of double-stranded RNA content distributions. J Histochem Cytochem 1980;28:663-9.
- Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 1998;98:794-9.
- Ren J. Leptin and hyperleptinemia: from friend to foe for cardiovascular function. J Endocrinol 2004;181:1-10.
- Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, Cioffi M, et al. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation 2002;105:804-9.
- Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002;105:2867-71.
- Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, et al. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 2002;106:3073-8.
- Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 2001;108:1429-37.
- Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003;108:1567-74.