
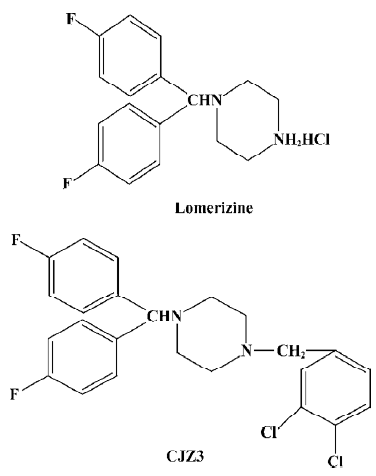
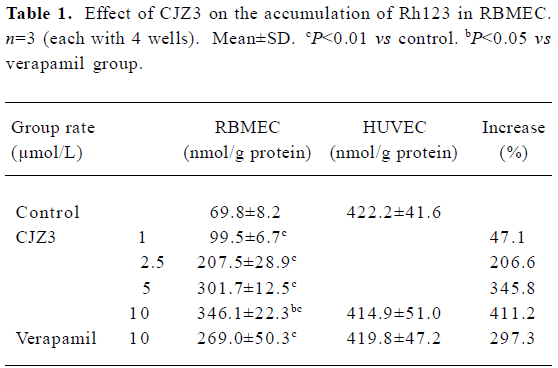
CJZ3, a lomerizine derivative, modulates P-glycoprotein function in rat brain microvessel endothelial cells1
Introduction
Treatment of cancer with chemotherapeutic drugs is frequently impaired or ineffective as a result of the acquired resistance of tumor cells. This phenomenon is termed multi-drug resistance (MDR), and is characterized by the over-expression of P-glycoprotein (P-gp) at the surface of cancer cells. P-gp is a 170 kDa protein that is encoded by an MDR gene, and is a membrane transporter of the ATP-binding cassette (ABC) superfamily. It acts as an energy-dependent drug efflux pump that prevents adequate intracellular accumulation of a broad range of cytotoxic drugs, including anthracyclines, vinca alkaloids, epipodophyllotoxins, and taxanes[1–3]. In addition, P-gp is also expressed in normal tissues such as the brain microvessel endothelial cells[4], which are responsible for the barrier property of the blood-brain barrier (BBB) and possess tight interendothelial cells junctions, but no pores or fenestrations. This barrier protects the brain against many exogenous toxins and sudden fluctuations in the levels of systemic substances. Hydrophobic agents such as vinblastine and doxorubicin, and lipid-soluble compounds, such as cyclosporin A, can not accumulate in the brain[5–7]. It has been proposed that P-gp expression in the brain microvessel endothelial cells is responsible for the extrusion of these compounds from the endothelial cells, thereby attenuating the accumulation of the drugs in the brain and possibly leading to the failure of therapy for brain disease. A variety of agents, such as verapamil (Ver) and cyclosporine A, have been reported to overcome MDR[8,9]. However, verapamil and cyclosporine A are used as antiarrhythmic and immunosuppressant drugs, respec-tively. Therefore, side effects are expected to arise when they are used as MDR-reversing agents. In light of these findings, the development of compounds that specifically inhibit P-gp function in cancer cells or the BBB might contribute to the treatment of cancers and central nervous system diseases. It has been reported that lomerizine, a calcium channel antagonist, has an inhibitory effect on the P-gp-mediated transport of daunorubicin[10]. The aim of the present study was to investigate the effects of CJZ3, a lomerizine derivative, on P-gp function in rat brain microvessel endothelial cells (RBMEC).
Materials and methods
Materials The human umbilical vein endothelial cells (HUVEC) were a gift from Prof Zhuang ZHANG (Beijing University of Chinese Medicine); the CJZ3 was obtained from Yun-gen XU (Novel Drug Research Centre, China Pharmaceutical University); rhodamine 123 (Rh123) and verapamil were purchased from Sigma (St Louis, MO, USA); fetal calf serum and Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1) medium were purchased from Gibco (USA); 1,1' -dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate acetylated low-density lipoprotein (DiI-acyl-LDL) were purchased from Sigma (St Louis, MO, USA). All other chemicals used in the experiments were commercial products of reagent grade.
Isolation and culture of RBMEC The RBMEC were isolated according the method of Abbott et al[11] with minor modifications. First, the cortexes were obtained from the brains of 10 Sprague-Dawley rats (d5; Jiangsu Province Animal Center) and placed in ice-cold phosphate-buffered saline (PBS). After the surface vessels and meninges were removed, the cortex gray matter was minced and incubated at 37 °C for 25 min in D’hanks containing 0.05% trypsin. Then, the samples were filtered with 150 µm nylon mesh. After centrifugation at 800×g for 5 min, the pellet was resuspended in PBS containing 20% bovine serum albumin (BSA) and centrifuged at 2000×g at 37 °C for 5 min. Fat, cell debris and myelin floating on the BSA were removed and the pellet containing the microvessels was resuspended and incubated at 37 °C for 30 min in PBS containing 0.1% collagenase II. The microvessels were finally collected by centrifugation at 800×g for 5 min, then the pellet was washed twice with PBS and cultured in DMEM/F12 (1:1) medium supplemented with 20% fetal bovine serum at 37 °C in a 5% CO2 humidified atmosphere.
Characterization of the RBMEC The RBMEC were washed twice with PBS, and incubated for 3 h at 37 °C with DiI-acyl-LDL (10 mg/mL), then washed 3 times with PBS and fixed in 4% paraformaldedyde in PBS. These cells were again washed 3 times with PBS, then mounted on glass slides in Fluoromount-G, and examined with a fluorescence microscope.
Rh123 intracellular accumulation assay The RBMEC were seeded at a density of 5×104/mL in 24-well plates. After the cells reached confluence, the cell monolayers were exposed to 5 µmol/L Rh123 in serum-free DMEM/F12 medium containing 1, 2.5, 5, or 10 µmol/L CJZ3 at 37 °C for 90 min. Verapamil, a P-gp inhibitor, was used as a positive control. After incubation, the medium was removed, and all monolayers were washed 3 times with ice-cold PBS and then dissolved in 1% Triton X-100. Rh123 fluorescence was determined by using a fluorescence spectrophotometer, and the concentration of Rh123 was measured from the fluorescence value by using an Rh123 standard curve. The amount of Rh123 in cell samples was normalized against the amount of protein in each sample as described previously[12,13].
Rh123 uptake assay The RBMEC were seeded at a density of 5×10 4/mL in 24-well plates, and then incubated with medium containing 5 µmol/L Rh123 in the presence of 0, 1, 2.5, 5, or 10 µmol/L CJZ3 for 10, 25, 45, 60, or 90 min at 37 °C in a humidified atmosphere of 5% CO2. After incubation, the medium was removed, and the cell monolayers were washed 3 times with ice-cold PBS and then dissolved in 1% Triton X-100. The concentration of Rh123 was determined as described for the accumulation assay. The rate constant (Kuptake) for uptake was obtained from fitting the data to Ft= (1–e–kt) Fss, where Ft is the amount of Rh123 at time t, Fss is the amount of Rh123 after 90 min. Graphs were plotted of the intracellular amount of Rh123 against time.
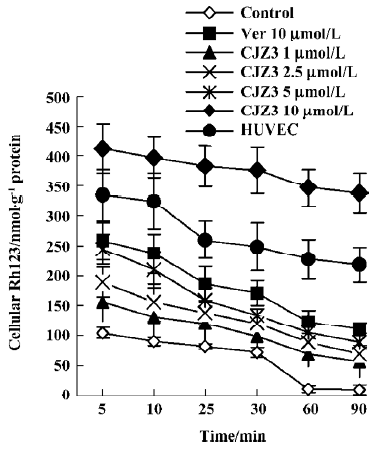
Rh123 efflux assay The RBMEC were first incubated in the medium containing 5 µmol/L Rh123 at 37 °C in a humidified atmosphere of 5% CO2 for 90 min, washed 3 times with ice-cold PBS, and then incubated in the presence of 0, 1, 2.5, 5, or 10 µmol/L CJZ3 at 37 °C for 5, 10, 25, 30, 60, or 90 min. The amount of intracellular Rh123 was determined as described for the accumulation assay. The rate constant (Kefflux) for the efflux caused by 10 µmol/L CJZ3 was obtained from fitting the data to Ft=e–ktFo, where Ft is the amount of Rh123 at time t. Graphs were plotted of the amount of intracellular Rh123 against time.
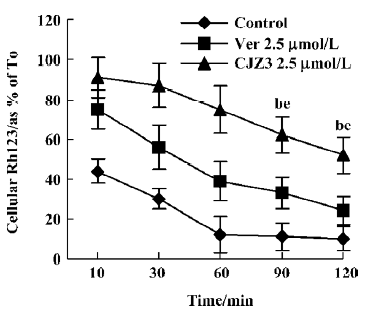
Persistence of activity The RBMEC were incubated in the medium containing 5 µmol/L Rh123 in the presence or absence of 2.5 µmol/L CJZ3 at 37 °C in a humidified atmosphere of 5% CO2 for 90 min, then washed with rhodamine-free and drug-free medium 3 times. At subsequent time points as indicated, the ability of the cells to accumulate Rh123 was measured. Graphs were plotted of the amount of intracellular Rh123 against time, where T0 represents the end of the modulator incubation phase.
Statistical analysis All data are presented as mean±SD and analyzed using analysis of variance (ANOVA) followed by the q test.
Results
Cell characterization Immunocytochemical labeling showed that the RBMEC labeled by DiI-acyl-LDL were taken approximately 91% of the total cultured cells of visual field.
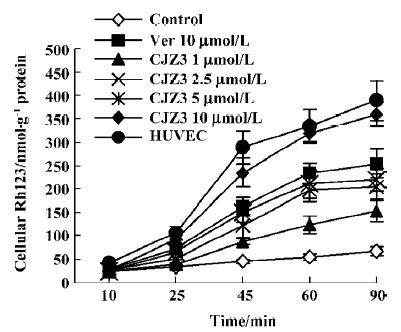
Effect of CJZ3 on intracellular accumulation of Rh123 After the RBMEC were incubated with Rh123 for 90 min in the presence of various concentrations of CJZ3, the amount of intracellular Rh123 was significantly increased compared with the control. CJZ3 caused Rh123 accumulation in a concentration-dependent manner. No such accumulation was found in HUVEC. The amount of intracellular Rh123 accumulated after treatment with 10 µmol/L CJZ3 was comparable to that caused by treatment with 10 µmol/L verapamil (Figure 1, Table 1).
Full table
Effect of CJZ3 on uptake of Rh123 When the RBMEC were incubated in the presence of 1, 2.5, 5, or 10 µmol/L CJZ3, they accumulated Rh123 in a time-dependent manner. The rate constant (Kuptake) of uptake was higher for the 10 µmol/L CJZ3-treated cells (0.060) than for the control cells (0.026) and the verapamil-treated cells (Figure 2, Table 2).
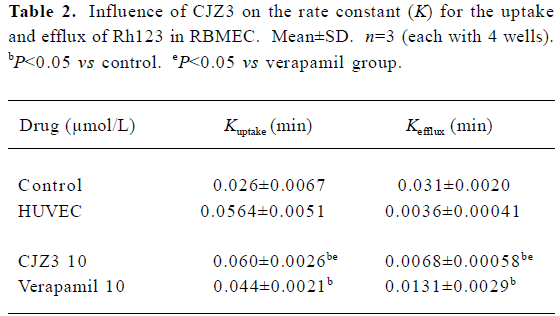
Full table
Inhibition of Rh123 efflux and persistence of activity Various concentrations of CJZ3 inhibited the efflux of Rh123 from the RBMEC, and the inhibitory effects remained for a period of 120 min after CJZ3 (2.5 µmol/L) was removed from the medium, suggesting that the inhibitory effects of CJZ3 on P-gp function are reversible and that the CJZ3 exerts its effects for longer than verapamil. The rate constant of the efflux was lower in 10 µmol/L CJZ3-treated cells (0.0068) than in control (0.031) and verapamil-treated cells (0.0131; Figures 3, 4, Table 2).
Discussion
The results of the present study indicate that primary cultured brain microvessel endothelial cells are valuable tools for the study of P-gp function in vitro. HUVEC, in which no detectable P-gp expression is found, is often used as a negative control[14,15]. The efflux of Rh123 fluorescent dye is known to be P-gp-dependent, and consequently has been used extensively to determine the efflux rate from cells expressing P-gp and to screen novel P-gp reversal agents[16]. In the present study, after DiI-acyl-LDL was incubated with the cultured cells, the RBMEC labeled by DiI-acyl-LDL were taken approximately 91% of the total cultured cells of visual field, indicating that the used method for the cultured cells was credible. The results of the present study also showed that after the RBMEC and HUVEC were exposed to Rh123, the amount of intracellular Rh123 in the RBMEC was lower than that in the HUVEC, and that the CJZ3 elevated the intracellular accumulation of Rh123 in RBMEC in a concentration-dependent manner (Table 1). No such accumulation was observed in HUVEC. The changes in cellular Rh123 concentration over the uptake time-course were the result of passive inward diffusion and outward active transport; however, the changes in cellular Rh123 over the efflux time-course were the result of outward diffusion and active transport, which were used to study P-gp function using continual and dynamic methods. These results show that CJZ3 significantly enhanced the uptake of Rh123 over the uptake phase and inhibited the efflux of Rh123 over the efflux phase, implying that CJZ3 had a more potent inhibitory effect on P-gp function than verapamil (Table 2, Figure 2, 3). Similar results for CJZ3 have been observed in doxorubicin-resistant human myelogenous leukemia (K562/DOX) cells (unpublished data). We also found that the ability of CJZ3 to cause the accumulation of Rh123 was greater than that of CJX1 and CJX2, the amlodipine derivatives studied previously[17]: the rate constant of uptake (Kuptake) was higher in 10 µmol/L CJZ3-treated cells (0.060) than in CJX1-treated cells (0.0508) and in CJX2-treated cells (0.0495), and the rate constant of efflux (Kefflux) was lower (0.0068) than in CJX1-treated cells (0.0097) and in CJX2-treated cells (0.0127), suggesting that CJZ3 also has a more potent inhibitory effect on P-gp function than CJX1 and CJX2.
Furthermore, the inhibitory effect of CJZ3 on BBB permeability was reversible (Figure 4), indicating that the P-gp functional activity in RBMEC that were pretreated with modulator could return to control levels after the removal of CJZ3 from the medium, which is significant for the recovery of the physiological function of P-gp. We also found that the duration of activity of CJZ3 was longer than that of verapamil. CJZ3 inhibited P-gp function in RBMEC for more than 120 min after a short exposure to 2.5 µmol/L CJZ3, which was subsequently removed from the incubation medium.
P-gp has 12 transmembrane domains contained in 2 homologous halves, and there are 2 ATP-binding cassette domains in each of the halves that catalyze ATP hydrolysis. P-gp also presents another 2 types of binding sites: one for transport (substrate-binding site) and one for modulation (modulator site). The various P-gp reversal agents may block substrate efflux by binding to similar substrate binding sites or to other modulator sites, which cause allosteric changes, resulting in inhibition of therapeutic drug binding or transport[18]. The present report is the first to present pharmacodynamic data for CJZ3. The exact mechanism by which CJZ3 interacts with P-gp will be investigated further in our laboratory.
The inhibitory effect of CJZ3 on P-gp function indicates that the compound may be able to reverse P-gp-mediated MDR. To determine if this is the case, we carried out research using doxorubicin-resistant human myelogenous leukemia (K562/DOX) cells, and the results will be published elsewhere. The effects of CJZ3 on P-gp function in vivo will be further studied in our laboratory.
In conclusion, CJZ3 had a potent in vitro effect on the inhibition of P-gp function and elevation of BBB permeability. It is possible that CJZ3 will be found to be an effective P-gp reversal agent. The present results revealed that it is possible to improve the therapeutic effects of drugs on central nervous diseases by administering CJZ3 concurrently.
References
- Germann UA. P-glycoprotein: a mediator of multidrug resistance in tumour cells. Eur J Cancer 1996;32A:927-44.
- Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 1993;62:385-427.
- Childs S, Ling V. The MDR superfamily of genes and its biological implications. In: DeVita VT, Hellman S, Rosenberg SA, editors. Important advances in oncology 1994. Philadelphia: J B Lippin-cott Company. 1994. p 21–36.
- Elsinga PH, Hendrikse NH, Bart J, Vaalburg W, van Waarde A. PET Studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS. Curr Pharm Des 2004;10:1493-503.
- Cisternino S, Rousselle C, Debray M, Scherrmann JM. In vivo saturation of the transport of vinblastine and colchicine by P-glycoprotein at the rat blood-brain barrier. Pharm Res 2003;20:1607-11.
- Zhao YL, Du J, Kanazawa H, Sugawara A, Takagi K, Kitaichi K, et al. Effect of endotoxin on doxorubicin transport across blood-brain barrier and P-glycoprotein function in mice. Eur J Pharmacol 2002;445:115-23.
- Sakata A, Tamai I, Kawazu K, Deguchi Y, Ohnishi T, Saheki A, et al. In vivo evidence for ATP-dependent and P-glycoprotein-mediated transport of cyclosporin A at the blood-brain barrier. Biochem Pharmacol 1994;48:1989-92.
- Twentyman PR. Modification of cytotoxic drug resistance by non-immuno-suppressive cyclosporins. Br J Cancer 1988;57:254-8.
- Aszalos A, Thompson K, Yin JJ, Ross DD. Combinations of P-glycoprotein blockers, verapamil, PSC833, and cremophor act differently on the multidrug resistance associated protein (MRP) and on P-glycoprotein (Pgp). Anticancer Res 1999;19:1053-64.
- Shiraki N, Hamada A, Ohmura T, Tokunaga J, Oyama N, Nakano M. Increase in doxorubicin cytotoxicity by inhibition of P-glycoprotein activity with lomerizine. Biol Pharm Bull 2001;24:555-7.
- Abbott NJ, Hughes CC, Revest PA, Greenwood J. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. J Cell Sci 1992;103:23-37.
- Fontaine M, Elmquist WF, Miller DW. Use of rhodamine 123 to examine the functional activity of P-glycoprotein in primary cultured brain microvessel endothelial cell monolayers. Life Sci 1996;59:1521-31.
- Sarver JG, Klis WA, Byers JP, Erhardt PW. Microplate screening of the differential effects of test agents on Hoechst 33342, rhodamine 123, and rhodamine 6G accumulation in breast cancer cells that overexpress P-glycoprotein. J Biomol Screen 2002;7:29-34.
- Zhu HJ, Liu GQ. Effect of E6, a novel calmodulin inhibitor, on activity of P-glycoprotein in purified primary cultured rat brain microvessel endothelial cells. Acta Pharmacol Sin 2003;24:1143-9.
- He L, Liu GQ. Effects of various principles from Chinese herbal medicine on rhodamine123 accumulation in brain capillary endothelial cells. Acta Pharmacol Sin 2002;23:591-6.
- Green LJ, Marder P, Slapak CA. Modulation by LY335979 of P-glycoprotein function in multidrug-resistant cell lines and human natural killer cells. Biochem Pharmacol 2001;61:1393-9.
- Ji BS, He L, Liu GQ. Modulation of P-glycoprotein function by amlodipine derivatives in brain microvessel endothelial cells of rats. Acta Pharmacol Sin 2005;26:166-70.
- Martin C, Berridge G, Higgins CF, Callaghan R. The multi-drug resistance reversal agent SR33557 and modulation of vinca alkaloid binding to P-glycoprotein by an allosteric interaction. Br J Pharmacol 1997;122:765-71.