
Regulation of nicotine-induced cyclooxygenase-2 protein expression in human gingival fibroblasts
Introduction
Cigarette smoking is a risk factor in the incidence and/or progression of periodontal disease[1,2]. Numerous studies have examined the effects of whole tobacco smoke as well as individual components on periodontal tissues, such as epithelium and connective tissue and on components of the inflammatory and immune response. A variety of studies have been carried out to assess the changes in levels of inflammatory and host response substances caused by nicotine, a major component of the particulate phase of all cigarette smoke. Previous studies have shown that nicotine is toxic to fibroblasts derived from periodontium through inhibiting cell viability, attachment, proliferation, and matrix protein synthesis[3−6]. Our recent studies have shown that nicotine can induce c-fos[7], cyclooxygenase-2 (COX-2)[8], and heme oxygenase-1[9] expression in fibroblasts derived from periodontium. However, the mechanism behind the nicotine-induced expression of c-fos, COX-2 or other signal proteins still remains to be elucidated.
COX is the rate-limiting enzyme that catalyzes the oxygenation of arachidonic acid to prostaglandin endoperoxidases which are converted enzymatically into prostaglandin and thromboxane A2. Two distinct isoforms of COX have been identified[10]. COX-1 is constitutively expressed at a low level in most tissues but in contrast, COX-2, the product of a related inducible gene, is absent from most tissues but is expressed in response to proliferative and inflammatory stimuli[11,12].
Recently, we showed that nicotine could induce COX-2 mRNA and protein expression in human gingival fibroblasts (HGF)[8]. As far as we know, little is known about whether chemical interactions can modulate nicotine-induced COX-2 expression. Antioxidants are substances that, when existing at low concentrations compared with those of the oxidizable substrate, significantly delay or prevent oxidation of that substrate[13]. To determine whether oxidative stress is important in the induction of COX-2 expression by nicotine, we pretreated cells with the glutathione (GSH) precursor, 2-oxothiazolidine-4-carboxylic acid (OTZ), to boost thiol levels, or buthionine sulfoximine (BSO) to deplete GSH. In addition, oxidative stress was found to activate extracellular signal-regulated protein kinase (ERK) signaling, which in turn has been implicated in the response to inflammation[14]. Furthermore, ERK inhibitor PD98059 was added to investigate the possible regulation mechanisms of nicotine-induced COX-2 expression. In the present study, we show that nicotine-induced COX-2 expression is augmented by oxidative stress and mediated by ERK signaling.
Materials and methods
Reagents Nicotine, BSO (a cellular GSH synthesis inhibitor), and OTZ (a precursor of cysteine) were purchased from Sigma (St Louis, MO, USA). PD98059 (an ERK inhibitor) was obtained from Promega (Madison, WI, USA). Mouse anti-human COX-2 monoclonal antibody was obtained from Transduction Laboratories (Lexington, KY, USA). Anti-ERK and anti-phospho-ERK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Tissue culture reagents were purchased from Gibco Laboratories (Grand Island, NY, USA). The final concentrations of nicotine, OTZ, BSO, and PD98059 used in this study were 5 mmol/L, 5 mmol/L, 50 µmol/L, and 10 µmol/L, respectively.
Cell culture The human biopsy materials used in this study were obtained according to the guidelines of the Oral Medicine Center and the Joint Research and Ethics Committee of the Chung Shan Medical University Hospital. Normal gingival tissue samples were obtained from 3 healthy individuals undergoing routine surgical crown lengthening, who had little if any evidence of inflammation, and who were receiving no systemic medication. HGF were cultured by using an explant technique as described previously[9]. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics (100 U/mL of penicillin, 100 µg/mL of streptomycin and 0.25 µg/mL of fungizone). Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Confluent cells were detached with 0.25% trypsin and 0.05% EDTA for 5 min, and aliquots of separated cells were subcultured. Cell cultures between the third and eighth passages were used.
Lactate dehydrogenase assessment Cellular toxicity was measured by the release of the cytoplasmic enzyme lactate dehydrogenase (LDH). Briefly, cells were seeded at an initial density of 5×105 in 60-mm culture dishes and allowed to attach for 24 h. The cells were then incubated with nicotine at concentrations of 0−20 mmol/L. Cells were detached with 0.25% trypsin and 0.05% ethylenediamine tetraacetic acid (EDTA) for 5 min. To measure LDH, 500 µL of the cell suspension was pelleted 24 h later. The LDH released into the supernatant was measured on the basis of reduced absorbance at 340 nm due to NADH consumption during the reaction of pyruvate to lactate catalyzed by LDH. The percentage of LDH leakage in response to nicotine as compared with untreated cells was calculated by the formula:
Chemical treatment Cells arrested in G0 by serum deprivation (0.5% FCS; 48 h) were used in the experiments[7,8]. Prior to treatment, the cells were washed with serum-free DMEM and immediately thereafter exposed to 5 mmol/L nicotine for the indicated incubation times. Cells were pre-exposed to OTZ or BSO for 24 h before the addition of nicotine. This protocol was necessary to allow changes in GSH levels to occur prior to exposure to nicotine. For COX-2 expression, cell lysates were collected at 4 h. For ERK and phosphor ERK expression, cell lysates were collected at 0, 0.5, 1, 2, 4, and 8 h.
Western blotting For Western blot analysis, cell lysates were collected as described previously[8,12]. Briefly, cells were solubilized with sodium dodecylsulfate (SDS) solubilization buffer [5 mmol/L EDTA, 1 mmol/L MgCl2, 50 mmol/L Tris-HCl(pH 7.5) and 0.5% Trition X-100, 2 mmol/L phenylmethylsulfonyl fluoride, and 1 mmol/L N-ethylmaleimide] for 30 min on ice. Then, cell lysates were centrifuged at 12 000×g at 4 °C and the protein concentrations were determined with Bradford reagent using bovine serum albumin (BSA) as standards. Equivalent amounts of total protein per sample of cell extracts were run on a 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and immediately transferred to nitrocellulose membranes. The membranes were blocked with phosphate-buffered saline (PBS) containing 3% BSA for 2 h, rinsed, and then incubated with primary antibodies anti-COX-2 (1:1000) or anti-ERK (1:1000) or antiphospho-ERK (1:1000) in PBS containing 0.05% Tween 20 for 2 h. After 3 washes with Tween 20 for 10 min, the membranes were incubated for 1 h with biotinylated secondary antibody diluted at 1:1000 in the same buffer, washed again as described earlier and treated with 1:1000 streptavidin-peroxidase solution for 30 min. After a series of washing steps, the reactions were developed using Diaminobenzidine (DAB, Zymed, South San Francisco, CA, USA). The intensities of the obtained bands were determined by a densitometer (AlphaImager 2000; Alpha Innotech, San Leandro, CA, USA). Each densitometric value was expressed as mean±SD.
Statistical analysis Three replicates of each concentration were performed in each test. All assays were repeated 3 times to ensure reproducibility. Statistical analysis was carried out by one-way analysis of variance (ANOVA). Tests of differences of the treatments were analyzed by Duncan’s test and a value of P<0.05 was considered statistically significant.
Results
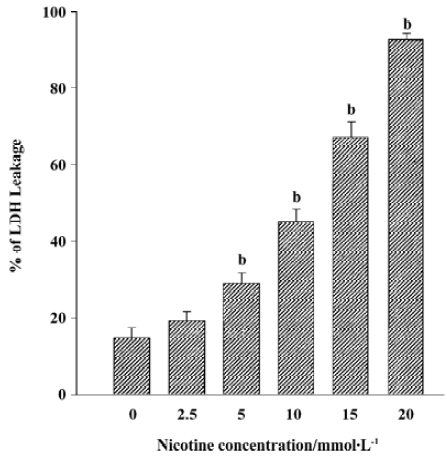
Leakage of LDH in HGF after exposure to nicotine for 24 h Nicotine was found to elevate LDH leakage in a dose-dependent manner (P<0.05). Nicotine was cytotoxic to HGF at concentrations of 5 mmol/L and above according to LDH assay (Figure 1). The amount of LDH leakage was approximately 32%, 45%, 68%, and 96% at concentrations of 5, 10, 15, and 20 mmol/L, respectively (Figure 1).
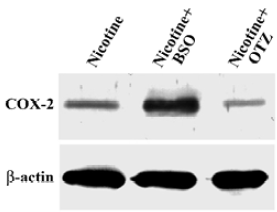
COX-2 protein expression in HGF induced by 5 mmol/L nicotine Nicotine upregulated COX-2 protein expression in HGF. In addition, pretreatment with OTZ decreased the nicotine-induced COX-2 protein level by approximately 60% (P<0.05). However, BSO enhanced the nicotine-induced COX-2 protein level by up to 3-fold (P<0.05; Figure 2).
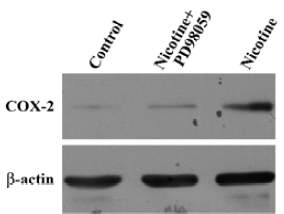
Effect of nicotine on ERK activation PD98059, a specific inhibitor of ERK, was used to block the activation of COX-2 expression. Treatment of HGF with PD98059 decreased the nicotine-induced COX-2 protein expression (Figure 3).
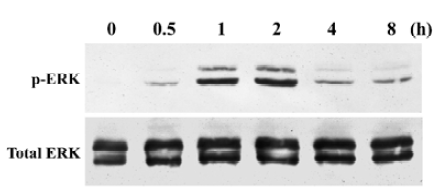
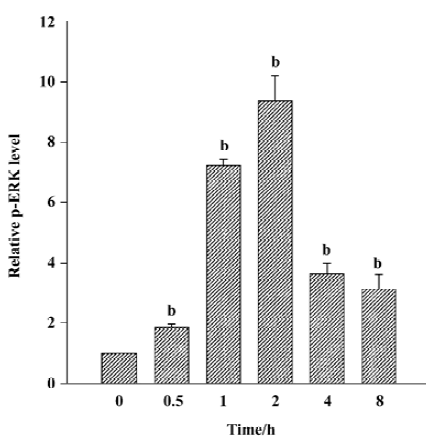
Nicotine induced ERK phosphorylation in a time-dependent manner (P<0.05, Figure 4), with maximal activity appearing 2 h after initiating exposure to nicotine. Amounts of ERK protein were unaffected by nicotine during the same time interval. The quantitative measurement of p-ERK protein was made by using the AlphaImager 2000 densitometer (Figure 5). The levels of the p-ERK increased approximately 1.9-, 7.1-, 9.4-, 3.7- and 3.1-fold after exposure to nicotine for 0.5, 1, 2, 4, and 8 h, respectively. However, cells resting in 0.5% FCS did not express detectable levels of p-ERK.
Discussion
Some of the components of tobacco products, for example nicotine, may contribute to the development of periodontal disease via direct effects on cell viability, host maintenance, and the cellular defense system. Normal fibroblast function is critical for the maintenance of periodontal tissues and for optimal wound healing responses. HGF were chosen for the present study due to their ready availability and particular culturing characters. Periodontal ligament fibroblasts would stimulate periodontal tissues better, but these cells are known to be similar to HGF[15]. In addition, Aukhil et al[16] have reported that only a very limited area of 200−400 µm of the periodontal ligament apical to the surgical wound actually participates as a source of regenerative cells. Thus, the effects of nicotine on HGF may have clinical significance.
In the present study, the cytotoxicity of nicotine was evaluated using an LDH leakage assay in HGFs. It was found that nicotine was toxic to HGF. Although the experimental conditions in the present study differed from those used in other studies, our results were in agreement with previous studies in that we found that nicotine was cytotoxic to human fibroblasts derived from periodontium[3–6]. It has been suggested that factors that inhibit the functions of HGF would also impair tissue repair and regeneration. Tobacco users may be more susceptible to destruction of the periodontium and less responsive to regeneration procedures during periodontal therapy.
COX-2 is an inducible enzyme that is believed to be responsible for prostaglandin synthesis at sites of inflammation. Data from our in vitro experiments confirmed our previous report that nicotine was capable of stimulating COX-2 protein expression in HGF[10]. Our results are also in agreement with those of Yan et al[17], who reported that benzo[α]pyrene, a polycyclic aromatic hydrocarbon found in tobacco smoke, induced the transcription of COX-2 in vascular smooth muscle cells, and of Anto et al[18], who demonstrated that cigarette smoke condensate induced COX-2 expression in human epithelial, lymophoid, and myeloid cells in vitro. Thus, one of the pathogenetic mechanisms of chronic periodontal inflammation may be the synthesis of COX-2 by resident cells in cigarette smokers.
The sulfphhydryl group containing tripeptide constitutes a first-defense intracellular antioxidant[19]. GSH plays a role in cellular protection from damage produced by free radicals and electrophiles. It is well-known BSO, a specific inhibitor of γ-glutamyl cysteine synthetase, inhibits GSH synthesis, is relatively non-toxic, and is quite efficient in decreasing intracellular GSH levels. OTZ, a precursor of cysteine, metabolically promotes GSH synthesis, increasing intracellular GSH levels by as much as 2 to 3 times the control level[20].
In the present study, we found that the extent of COX-2 induction by nicotine in HGF depended upon the antioxidant potential of the HGF, being strongly enhanced when GSH was depleted by pretreatment with BSO, an inhibitor of GSH synthesis, and strongly diminished by treatment with OTZ, a GSH synthesizer. These data suggest that the oxidant effects of nicotine mediate the induction of COX-2 in HGF, a finding consistent with previous reports that oxidative stress can induce COX-2[17,21]. These results suggest that thiol pools may act as an intracellular buffer against nicotine-induced COX-2 expression.
COX-2 expression has been linked with the activation of the ERK signaling transduction pathway[22,23]. To further investigate the mechanism of nicotine-induced signaling proteins, Western blot analysis of the phosphorylated proteins was performed. Phosphorylation of ERK was increased, whereas no increase was observed in ERK. COX-2 induction via preferential activation of ERK has been associated with resistance to oxidative stress in smooth muscle cells[17]. Recently, similar results were found by Huang et al[24] who reported that nicotine could directly induce p-ERK expression in human osteosarcoma cells. Taken together, these data provide the first evidence that activation of ERK pathway is directly involved in the nicotine-induced COX-2 expression.
Data from the present study indicate that the effect of nicotine in HGF is through activation of the COX-2 pathway. COX-2 expression is critically dependent on cellular thiol levels allowing nicotine to interfere with specific target molecules resulting in the expression of COX-2. COX-2 expression is directly involved in the phosphorylation of ERK. Thus, nicotine-induced COX-2 expression is augmented by oxidative stress and mediated by ERK signaling. Understanding the cellular processes involved and the molecular mechanisms responsible for COX-2 induction by nicotine may aid in the development of more effective treatments for cigarette smoking-associated periodontal disease. Factors that induce glutathione synthesis or ERK inhibitors suppress COX-2 expression and may therefore be clinically useful agents, in combination with standard treatment modalities, in the treatment of COX-2-mediated cigarette smoking-associated periodontal destruction.
References
- Haber J, Wattles J, Crowley M, Mandell R, Joshipura K, Kent RL. Evidence for cigarette smoking as a major risk factor for periodontitis. J Periodontol 1993;64:16-23.
- Bergstrom J, Eliasson SD, Dock J. Exposure to tobacco smoking and periodontal health. J Clin Periodontol 2000;27:61-8.
- Tipton DA, Dabbous MKh. Effects of nicotine on proliferation and extracellular matrix production of human gingival fibroblasts in vitro. J Periodontol 1995;66:1056-64.
- Giannopoulou C, Geinoz A, Cimason G. Effects of nicotine on periodontal ligament fibroblasts in vitro. J Clin Periodontol 1999;26:49-55.
- Chang YC, Lii CK, Tai KW, Chou MY. Adverse effects of arecoline and nicotine on human periodontal ligament fibroblasts in vitro. J Clin Periodontol 2001;28:277-82.
- Chang YC, Huang FM, Tai KW, Yang LC, Chou MY. Mechanisms of cytotoxicity of nicotine in human periodontal ligament fibroblast cultures in vitro. J Periodontal Res 2002;37:279-85.
- Chang YC, Hsieh YS, Lii CK, Huang FM, Tai KW, Chou MY. Induction of c-fos expression by nicotine in human periodontal ligament fibroblasts is related to cellular thiol levels. J Periodontal Res 2003;38:44-50.
- Chang YC, Tsai CH, Yang SH, Liu CM, Chou MY. Induction of cyclooxygenase-2 mRNA and protein expression in human gingival fibroblasts stimulated with nicotine. J Periodontal Res 2003;38:496-501.
- Chang YC, Lai CC, Lin LF, Ni WF, Tsai CH. The upregulation of heme oxygenase-1 expression in human gingival fibroblasts stimulated with nicotine. J Periodontal Res 2005;40:252-7.
- Maier JAM, Hla T, Maiciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem 1990;265:10805-8.
- Chang YC, Yang SF, Huang FM, Liu CM, Tai KW, Hsieh YS. Proinflammatory cytokines induce cyclooxygenase-2 mRNA and protein expression in human pulp cell cultures. J Endod 2003;29:201-4.
- Chang YC, Huang FM, Yang SF, Liu CM, Lai CC, Chan Y, et al. Induction of cyclooxygenase-2 mRNA and protein expression in human pulp cells stimulated with black-pigmented Bacteroides. J Endod 2003;29:240-3.
- Halliwell B. Antioxidants: the basic what they are and how to evaluate them. In: Antioxidants in disease mechanisms and therapy. Advances in pharmacology; volume 38. San Diego: Academic Press, 1997. p 3−20.
- Adderly SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem 1999;274:5038-46.
- Hou LT, Yaeger JA. Cloning and characterization of human gingival and periodontal ligament fibroblasts. J Periodontol 1993;64:1209-18.
- Aukhil I, Simpson DM, Suggs C, Pettersson E. In vivo differentiation of progenitor cells of the periodontal ligament. J Clin Periodontol 1986;13:862-8.
- Yan Z, Subbaramaiah K, Camilli T, Zhang F, Tanabe T, McCaffrey TA, et al. Benzo[α]pyrene induces the transcription of cyclo-oxygenase-2 in vascular smooth muscle cells. J Biol Chem 2000;275:4949-55.
- Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwa BB. Cigarette smoke condensate activates nuclear transcription factor-κB through phosphorylation and degradation of IκBα: correlation with induction of cyclooxygenase-2. Carcinogenesis 2002;23:1511-8.
- Meister A. Selective modification of glutathione metabolism. Science 1983;220:472-7.
- Williamson JM, Boettcher B, Meister A. Intracellular cysteine delivery system that protects against toxicity by promoting glutathione synthesis. Proc Natl Acad Sci USA 1982;79:6246-9.
- Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipo-polysaccharide. J Clin Invest 1995;95:1669-75.
- Guan Z, Buckman SY, Miller BW, Springer LD, Morrison AR. Interleukin-1beta-induced cyclooxygenase-2 expression requires activation of both c-Jun NH2-terminal kinase and p38 MAPK signal pathways in rat renal mesangial cells. J Biol Chem 1998;273:28670-6.
- Subbaramaiah K, Chung WJ, Dannenberg AJ. Ceramide regulates the transcription of cyclooxygenase-2. Evidence for involvement of extracellular signal-regulated kinase/c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways. J Biol Chem 1998;273:32943-9.
- Huang CY, Chen JH, Tsai CH, Kuo WW, Liu JY, Chang YC. Regulation of extracellular signal-regulated protein kinase signaling in human osteosarcoma cells stimulated with nicotine. J Periodontal Res 2005;40:176-81.