
A long-form α-neurotoxin from cobra venom produces potent opioid-independent analgesia1
Introduction
There have been numerous anecdotal reports of venoms being employed as analgesics in attempts to relieve severe pain associated with cancer, immune dysfunction and viral infection. Current methods to treat severe pain mainly comprise opiate-based products that have short-lived activity and the potential to produce dependence. New methods to relieve chronic pain are of great interest, and venoms are rich in peptides with the potential to modulate chronic pain. This aspect is highlighted by the recent approval of ziconitide (conotoxin SNX111) for the treatment of chronic pain.
Snake venoms have demonstrated antinociceptive activity, and certain isolated neurotoxins have demonstrated significant analgesia in animal models. Cobra venoms contain high levels of neurotoxins that target nicotinic acetylcholine receptors (NAChR). Cobrotoxin (CT)[1], a short-chain postsynaptic α-neurotoxin isolated usually from Naja naja atra, is reported to have analgesic activity[2,3] and is commercially available in China for this purpose. CT is considered to be a specific ligand for the muscle-based alpha1 subtype of the NAChR, although it produces strong, apparently centrally-mediated analgesic effects through an opiate-independent mechanism[3]. The recent demonstration that the analgesics ketamine and tramadol inhibit nicotinic currents carried by alpha7 receptors expressed in Xenopus oocytes[4] implicates the alpha7 receptor subtype. It was concluded that ketamine inhibits the presynaptic nicotinic receptors responsible for facilitating neurotransmitter release, as well as the direct ligand-gated inward current[4]. Ketamine was found to inhibit the nicotine-evoked presynaptic facilitation of glutamate release[5]. It has been reported that alpha-bungarotoxin can block the effects of ketamine. Alpha-cobratoxin (CTX), a long-chain neurotoxin and homolog of alpha-bungarotoxin, is a high-affinity ligand for the alpha7 subtype[6–8], whose localization is reported to be mainly pre-synaptic in the peripheral nervous system, although with high-affinity sites within the brain but not in the spinal cord. It is also known that alpha7 subtypes can conduct Ca2+ ions, thereby impacting directly on neurotransmitter release[9]. However, it is unknown if such long-chain neurotoxins can reach the CNS and exert antinociceptive activity.
The antinociceptive effects of neurotoxins from snake venoms other than that of Naja naja atra have been reported[10]. Crotamine (ip, sc), one of the main components in the venom of Crotalus durissus terrificus, produced analgesic effects[11]. Recently, we have demonstrated that crotoxin, another major neurotoxin of Crotalus durissus terrificus venom, in addition to its antitumor effects[12,13], has separate antinociceptive effects (HL Zhang et al, unpublished data) and demonstrates synergism with acetylsalicylic acid. It has been associated with reduced pain in patients with solid tumors in a phase I clinical trials[14]. These studies suggest that snake neurotoxins could provide the basis for new treatments to combat pain. The present study reports that CTX from the Thailand cobra, Naja kaouthia, provides antinociception in rodent pain models and that this activity is independent of the opioid pathway.
Materials and methods
Animals Male and female Kunming mice weighing 18–22 g and Sprague-Dawley rats weighing 200–250 g were purchased from the Center for Medical Experimental Animals, Soochow University, China (grade 2, certification N
Drugs and drug administration CTX was supplied by ReceptoPharm (Plantation, Florida, USA). Naloxone hydrochloride, acetylsalicylic acid and atropine sulfate were purchased from Sigma (St Louis, MO, USA).
Intra-cerebral ventricle injection The injection of CTX was performed as previously described[15]. Briefly, mice were anesthetized with methoxyflurane and a small incision was made on the skull to access the bregma. Mice were randomly divided into 2 groups (n=10 in each group): the saline control and 4.5 µg/kg CTX groups. CTX was administered in a volume of 5 µL normal saline through a puncture point 2 mm lateral to the bregma, using a 10-µL Hamilton syringe with a truncated 27 gauge needle so that it penetrated into the brain 3 mm from the top of the skull. The impact on the pain threshold was measured using the mouse acetic acid-writhing test 1 h after icv CTX or saline administration. To confirm that the drugs were administered into the cerebral ventricle, several mice were injected with 5 µL of diluted blue ink, and after the mice were killed, their brains were examined macroscopically after sectioning. The accuracy of the injection technique was found to be good, with 95% of injections being correctly located.
Periaqueductal gray cannulation and microinjection Under chloral hydrate anesthesia, a guide cannula (stainless steel tube of 0.3 mm OD) was stereotaxically implanted and positioned at 3 mm above the target area in rats. The coordinates were: AP, 6.3 mm posterior to the bregma; H, 5.8 mm below the dura mater; and L, 0.5 mm lateral to the midline[16,17]. The cannula was anchored on the skull with dental acrylic cement. Rats were housed individually in cages with food and water provided ad libitum and given 1 week to recover from surgery. One microliter of the drug solution (5 µg/kg) was injected over 5 min using the syringe cannula, which projected 3 mm beyond the tip of the guide cannula. The injection cannula was left in place for an additional 5 min to minimize the backflow of the drug. The delivery of drug to the periaqueductal gray (PAG) was also verified by injecting 1 µL ink through the guide cannula upon completing the observation.
Analgesic assessments
Hot-plate assay In the hot-plate test, female mice and rats were placed on a hot plate with the temperature setting controlled at 55 °C. The latency period required for mice and rats to lick their hind paws was recorded as the pain threshold. The baseline pain threshold was obtained by averaging the values of 2 measurements before drug or placebo administra-tion. Mice with 5–20 s or rats with 5–15 s pain thresholds were used in the experiments. Following drug administration, the pain threshold was determined as described and cut-off times of 60 s in mice and 30 s in rats were used in order to minimize injurious effects to the animals.
To assess the effects of CTX on pain response in the hot-plate test, female mice were randomly divided into 4 groups (n=10 in each group): normal saline (NS) was used as a control (group 1), and CTX was administered at doses of 30, 45, 68 µg/kg (groups 2–4). The pain threshold was measured at 1 to 24 h after drug (ip) administration.
Acetic acid writhing assay In the acetic acid writhing test, 10 min after the administration of 0.1 mL/10 g (ip) 1% acetic acid solution to mice, the number of writhing movements was counted from 10 min to 20 min after acetic acid injection.
The effects of CTX on pain response in the acetic acid writhing test was measured by randomly dividing mice into 4 groups (n=10 in each group), using saline as the control and CTX at 30, 45, and 68 µg/kg as for the hot-plate test. The pain threshold was measured 3 h after drug administration.
Assessment of effects of atropine and naloxone on CTX-induced analgesia The influence of atropine on CTX-induced analgesia in these models was also studied. In each assay, 4 groups of mice (n=10 in each group) were used: group 1, normal saline (NS) control; group 2, CTX at 45 mg/kg; group 3, atropine (Atr) at 0.5 mg/kg (in hot-plate test) or 10 mg/kg (in acetic acid writhing test); and group 4, CTX plus atropine (CTX+Atr) at the doses described for the individual assays. Atropine at a dose of 0.5 mg/kg (im) or 10 mg/kg (ip), or NS was administered 1.5 h after CTX. The pain threshold was determined 1.5 h after atropine administration.
The effects of naloxone on CTX-induced analgesia were assessed in the hot-plate test. Female mice were randomly divided into 6 groups (n=10 in each group): normal saline (NS) control, 45 µg/kg CTX, 1 or 5 mg/kg naloxone (Nal), and CTX plus 1 mg/kg or 5 mg/kg naloxone (CTX+Nal 1 mg/kg and CTX+Nal 5 mg/kg, respectively) as per the individual assays. Naloxone or NS (ip) was administered 2.5 h after CTX. The pain threshold was determined 30 min after naloxone administration.
Study of analgesic combinations The impact of acetylsalicylic acid (ASA) on the analgesia induced by CTX was investigated using the hot-plate test. In a manner similar to the assays described earlier, female mice were randomly divided into 4 groups (n=10 in each group): 1, normal saline (NS) control; 2, 45 µg/kg CTX; 3, 300 mg/kg ASA; and 4, CTX+ASA at the levels described previously. ASA (300 mg/kg, ip) was administered 2 h after cobratoxin (45 µg/kg). The pain threshold was determined 1 h after ASA administration.
Effects of CTX on locomotor function Locomotor activity in mice was measured using an Animex activity Type S meter (LKB, Farad, Sweden) with the setting at maximum sensitivity. Every movement by the mice was recorded automatically by the instrument. On the day of the experiment, the mice were treated with the highest level of CTX (68 µg/kg) or saline. The locomotor activity of mice was then observed for 15 min at 1, 2, and 3 h after ip injection of CTX or control.
Data analysis Data are expressed as mean±SD. Statistical significance of differences was determined by one-way analysis of variance (ANOVA).
Results
Effects of CTX on pain responses in mice and rats CTX at 30, 45, or 68 µg/kg (ip) produced a dose-dependent prolongation of the latency for mice to respond to pain stimulation induced by heat. The analgesic effect of CTX appeared at 2 h and peaked at 3 h after drug administration (Figure 1A). The ED50 of the antinociceptive effect of CTX was 57.6 µg/kg (35.32–93.93, 95% confidence limit) in the hot-plate test. Similarly, CTX produced a dose-dependent inhibition of the writhing response to acetic acid administration in mice (Figure 1B). The ED50 of the antinociceptive effect of CTX was 54.04 µg/kg (41.04–71.16, 95% confidence limit) for the acetic acid writhing test. The efficacy of CTX in analgesia was comparable to that of CT (ED50 was 63.10 µg/kg as assessed in the hot-plate model, or 55.86 µg/kg as assessed in the acetic acid writhing model in our parallel studies; data not shown). The analgesic effects disappeared 5 h after CTX (data not shown).

Analgesic actions of centrally administered CTX In mice, intra-cerebral ventricular (icv) administration of CTX (4.5 µg/kg), 1/12th of the systemic dose of CTX, significantly reduced the writhing response induced by acetic acid (P<0.05), indicating that icv injection of CTX was effective (Figure 2A). In the rat hot-plate test, administration of CTX (4.5 µg/kg) to PAG, 1/12th of a mouse systemic dose of CTX, did not produce a significant analgesic action (Figure 2B).

Influence of the antagonists atropine and naloxone In the hot-plate and acetic acid writhing test in mice, 0.5 mg/kg (im) or 10 mg/kg (ip) atropine alone had no significant effect on the pain threshold. In the hot-plate test, both CTX and CTX combined with a small dose of atropine (0.5 mg/kg) produced significant analgesia (P<0.05). There was no significant difference between the 2 groups (Figure 3A). How-ever, in the acetic acid writhing test, CTX produced marked analgesia, whereas CTX combined with a larger dose of atropine (10 mg/kg) did not produce significant analgesic action (Figure 3B), indicating that a large dose of atropine could antagonize the effects mediated by CTX.
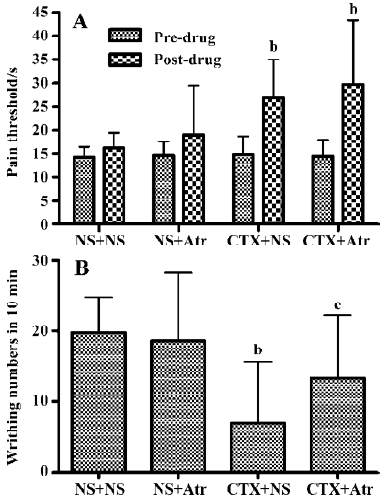
Naloxone at doses of 1 and 5 mg/kg (ip) had no significant influence on the pain threshold in the hot-plate assay. Both CTX (45 µg/kg) and CTX combined with naloxone produced similar analgesic effects (P<0.05). There was no significant difference between these groups (Figure 4).

CTX combined with ASA In the hot-plate test, CTX (45 µg/kg), ASA (300 mg/kg) and CTX combined with ASA produced marked analgesia. There was no significant difference among these 3 groups (Figure 5), providing evidence that no antagonism or synergism between the 2 products exists.

Locomotor effects of CTX To rule out the possibility that the effects of CTX on pain response were caused by an impairment of motor activity, mice pretreated with the highest dose of CTX (68 µg/kg, ip) were evaluated for spontaneous mobility 1, 2, and 3 h after drug administration with an Animex apparatus. The spontaneous mobility of mice did not change with treatment at the highest dose of CTX (68 µg/kg) used in the study as compared with saline-treated mice (Figure 6).

Discussion
It is known that α-neurotoxins such as CTX and CT have neurotoxic effects because they block nicotinic receptors at the neuromuscular junction[18], more specifically at the diaphragm. Despite this fact, cobra neurotoxins have been employed as analgesics for decades[2], and early studies in animal models confirmed that CT from Naja naja atra had antinociceptive properties[3]. In our present study CTX, a long-chain postsynaptic α-neurotoxin from Naja kaouthia, produced potent antinociceptive effects in both the hot-plate test and the acetic acid writhing test in mice when administered via icv and ip routes. The analgesic effects of CTX appeared 2 h following administration and reached a peak 3 h later. The ED50 of the antinociceptive effects of CTX was 57.60 µg/kg in the hot-plate test and 54.04 mg/kg in the acetic acid writhing test in mice. The observations made in the present study for CTX are very similar to those reported for CT, with the exception that atropine can inhibit the activity of CT at lower doses than observed for CTX. The present study showed that a large dose of atropine (10 mg/kg, ip) could antagonize the analgesic effects of CTX in the acetic acid writhing test in mice, suggesting that the analgesia induced by this toxin may be mediated by the central cholinergic system. Our study showed that naloxone had no effect on the analgesia induced by CTX, implying that the central endogenous opioid peptidergic system is not involved. This property is similar to that described for CT[3]. These results suggest that CTX, despite its higher toxicity/receptor affinity, is as effective as CT in the induction of analgesia and that the effect is not associated with the neurotoxic properties of the proteins. Studies using crotoxin have suggested a potent synergism with ASA (unpublished data), so CTX was assessed in combination with ASA, but no meaningful synergistic effects were noted.
Systemic and icv administration was effective for both neurotoxins, suggesting that the site of analgesic action of the neurotoxins may be, at least partially, in the central nervous system. The PAG matter has long been suggested to play a critical role in central pain inhibitory systems and morphine-mediated analgesia[19,20]. However, the present results demonstrated that microinjection of CTX at the same dose as that of icv (4.5 µg/kg) into the PAG did not result in significant analgesia, suggesting that the PAG is unlikely to be an active site. Constant administration of small doses of CT has been reported to increase the Leuenkephalin content in the hypothalamus, striatum and midbrain, and increase the Met-enkephalin content in the hypothalamus and midbrain, especially the thalamencephalon[21]. In general, the onset of activity averaged 3 h after administration, whether administered by icv or ip, and in the case of CT persisted for more than 6 h. This activity is not consistent with the known pharmacokinetics of these neurotoxins, and such neurotoxins show little accumulation in the central nervous system[22]. It is therefore not possible to rule out the possibility that CTX also exerts its antinociceptive effects peripherally or, as proposed by Chen and Robinson[3], that these neurotoxins act through a second messenger system. These postulations could serve to explain the time course of the measured responses.
It has been demonstrated that activation of the central cholinergic system produces antinociception in animals[15–20]. Activation of cholinergic pathways by nicotine and nicotinic agonists has been shown to elicit antinociceptive effects in a variety of species and pain tests. During the 1990s, the discovery of the antinociceptive properties of the potent nAChR agonist epibatidine in rodents sparked interest in the analgesic potential of this class of compounds[23]. The identification of considerable nAChR diversity suggested that the toxicities and therapeutic actions of the compound might be mediated by distinct receptor subtypes and, accordingly, epibatidine and its derivatives were used to identify nAChR with mainly alpha4 receptors, although receptors with alpha3 were also sensitive to these com-pounds. The involvement of alpha7 nicotinic receptors in nicotinic analgesia has been assessed through spinal (it) and icv administration in mice. Dose-dependent antinocicep-tive effects were seen with the alpha7 agonist choline after spinal and supraspinal injection using the tail-flick test[24]. Furthermore, alpha7 antagonists MLA and alpha-bungaro-toxin significantly blocked the effects of choline. These studies suggested that activation of alpha7 receptors in the central nervous system elicits antinociceptive effects in an acute thermal pain model. Nicotine’s analgesic effects are now believed to arise as a result of the desensitized state of the receptor following its activation[24]. In contrast to the roles of agonists discussed earlier, our own studies and those of others suggest that nicotinic antagonists may also have a role in pain relief. CTX and its homologue, alphabungaro-toxin, preferentially target the alpha7 and alpha1 nAchR in nerve and muscle tissue, respectively, and function by preventing the activation of such acetylcholine receptors in pre- and post-synaptic membranes. The lack of effect of atropine on the analgesic action of CTX in the present study could due to the low dose of atropine used. However, in a mouse hot-plate pain model, atropine at a dose larger than 0.5 mg/mg elevated the pain threshold. We thus tested a high dose of atropine (10 mg/kg) in the acetic acid pain model. We found that 10 mg/kg atropine had no effect on acetic acid-induced pain, but antagonized the analgesic action of CTX.
Cobra neurotoxins, mainly CT, have been employed clinically for the relief of pain. Derivatives of CTX are also being investigated in clinical applications[25]. The ease of administration provides such toxins with some advantages over conotoxin-based analgesics, which are administered intra-thecally[26]. Additionally, research is emerging that cobro-toxin can substitute for morphine and suppress the effects of morphine withdrawal[21]. The results of the present study suggest that CTX has analgesic effects and that the site of analgesic action may be the central nervous system, but the PAG is unlikely to be involved in the mechanism of action. The central cholinergic system appears to be involved in the antinociceptive action of CTX, although details of the mechanism remain unclear, and further studies on the relationship between central and peripheral cholinergic activity for the analgesic effect of CTX are necessary. These data may provide support for the development of a new analgesic drug based on CTX.
References
- Yang CC. Cobrotoxin: structure and function. J Nat Toxins 1999;8:221-33.
- Grasset E. The cobra neurotoxin; pharmacology and clinical applications in the treatment of pain. Med Hyg (Geneve) 1952;10:55-8.
- Chen R, Robinson SE. Effect of cholinergic manipulations on the analgesic response to cobrotoxin in mice. Life Sci 1990;47:1949-54.
- Shiraishi M, Minami K, Uezono Y, Yanagihara N, Shigematsu A, Shibuya I. Inhibitory effects of Tramadol on nicotinic acetylcholine receptors in adrenal chromaffin cells and in Xenopus oocytes expressing alpha7 receptors. Br J Pharmacol 2002;136:207-16.
- Irnaten M, Wang J, Venkatesan P, Evans CK, Chang KS, Andresen MC, et al. Ketamine inhibits presynaptic and postsynaptic nicotinic excitation of identified cardiac parasympathetic neurons in nucleus ambiguus. Anesthesiology 2002;96:667-74.
- Lukas RJ. Diversity and patterns of regulation of nicotinic receptor subtypes. Ann NY Acad Sci 1955;757:153-8.
- Servent D, Anti-Delbeke S, Gaillard C, Corringer PJ, Changeux JP, Menenz A. Molecular characterization of the specificity of interactions of various neurotoxins on two distinct nicotinic acetylcholine receptors. Eur J Pharmacol 2000;393:197-204.
- Dajas-Bailador F, Costa G, Dajas F, Emmett S. Effects of α-bungarotoxin, α-cobratoxin and fasciculin on the nicotine-evoked release of dopamine in the rat striatum in vivo. Neurochem Intl 1998;33:307-12.
- Lena C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci 1997;17:576-85.
- Pu XC, Wong PT, Gopalakrishnakone P. A novel analgesic toxin (hannalgesin) from the venom of king cobra (Ophiophagus hannah). Toxicon 1995;33:1425-31.
- Adriana CM, Andreimar MS, Silvia HAE, Vitor MF, Lewis G, Sergio Z, et al. The analgesic activity of crotamine, a neurotoxin from Crotalus durissus terrificus (South American rattlesnake) venom: a biochemical and pharmacological study. Toxicon 1998;36:1927-37.
- Rudd CJ, Viskatis LJ, Vidal JC, Etcheverry MA. In vitro comparison of cytotoxic effects of crotoxin against three human tumors and normal human epidermal keratinocyte cell line. Invest New Drugs 1994;12:183-4.
- Corin RE, Viskatis LJ, Vidal JC, Etcheverry MA. Cytotoxicity of Crotoxin on murine erythroleukemia cells in vitro. Invest New Drugs 1993;11:11-5.
- Cura JE, Blanzaco DP, Brisson C, Cura MA, Cabrol R, Larrateguy L, et al. Phase I and pharmacokinetics study of crotoxin (cytotoxin PLA2, NSC-624244) in patients with advanced cancer. Clin Cancer Res 2002;8:1033-41.
- Pedigo NW. Dewey Wl, Harris LS. Determination and characterization of the antinociceptive activity of intraventricularly administered acetylcholine in mice. J Pharmacol Exp Ther 1975;193:845-52.
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2nd ed. New York: Academic Press; 1986.
- Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain sites sensitive to the direct application effects with the periaqueductal gray. Brain Res 1976;114:1183-8.
- Karlsson E. Chemistry of protein toxins in snake venoms. In: Lee CY, editor. Handbook of experimental pharmacology. New York: Springer-Verlag; 1979. p 159–83.
- Jensen TS, Yaksh TL. Comparison of antinococeptive action of morphine in the periaqueductal gray, medial and puramedial medulla in rat. Brain Res 1986;363:99-113.
- Harris LS, Dewey WL. Role of cholinergic systems in the central action of narcotic agonists and antagonists. In: Kosterlitz HW, Collier HOJ, Villareal JE, editors. Agonist and antagonist actions of narcotic analgesic drugs. London: Macmillan 1972. p 198–206.
- Xiong Y, Wand W, Pu X, Song J. Preliminary study on the mechanism of using snake venoms to substitute for morphine. Toxicon 1992;30:567.
- Tseng LF, Chiu TH, Lee CY. Absorption and distribution of I-labeled cobra venom and its purified toxins. Toxicol Appl Pharmacol 1968;12:526-35.
- Decker MW, Meyer MD, Sullivan JP. The therapeutic potential of nicotinic acetylcholine receptor agonists for pain control. Expert Opin Investig Drugs 2001;10:1819-30.
- Damaj MI, Meyer EM, Martin BR. The antinociceptive effects of alpha7 nicotinic agonists in an acute pain model. Neuropharmacology 2000;39:2785-91.
- Mundy HR, Jones SJ, Hobart JC, Hanna MG, Lee PJ. A randomized controlled study of modified cobratoxin in adrenomyelo-neuropathy. Neurology 2003;61:528-30.
- Smith MT, Cabot PJ, Ross FB, Robertson AD, Lewis RJ. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain 2002;96:119-27.