
JNK pathway: diseases and therapeutic potential1
Introduction
Cells have to respond to environment stimuli properly to survive. These stimuli include physical stimulation such as heat, pH variation, radiation, redox, osmolarity, and chemical stimulation such as growth factors, cytokines, hormones, alterations in nutrient conditions, as well as other environment stresses[1]. These stimuli control many aspects of complex cellular processes, including the regulation of gene expression, cell survival, growth, differentiation and death. In these processes, many signal transduction pathways cooperate to relay, amplify, and integrate a diverse range of extracellular stimuli. The mitogen-activated protein kinase (MAPK) signaling pathway is one of the major signaling systems that transduce extracellular signals into cells[1,2] (Figure 1).
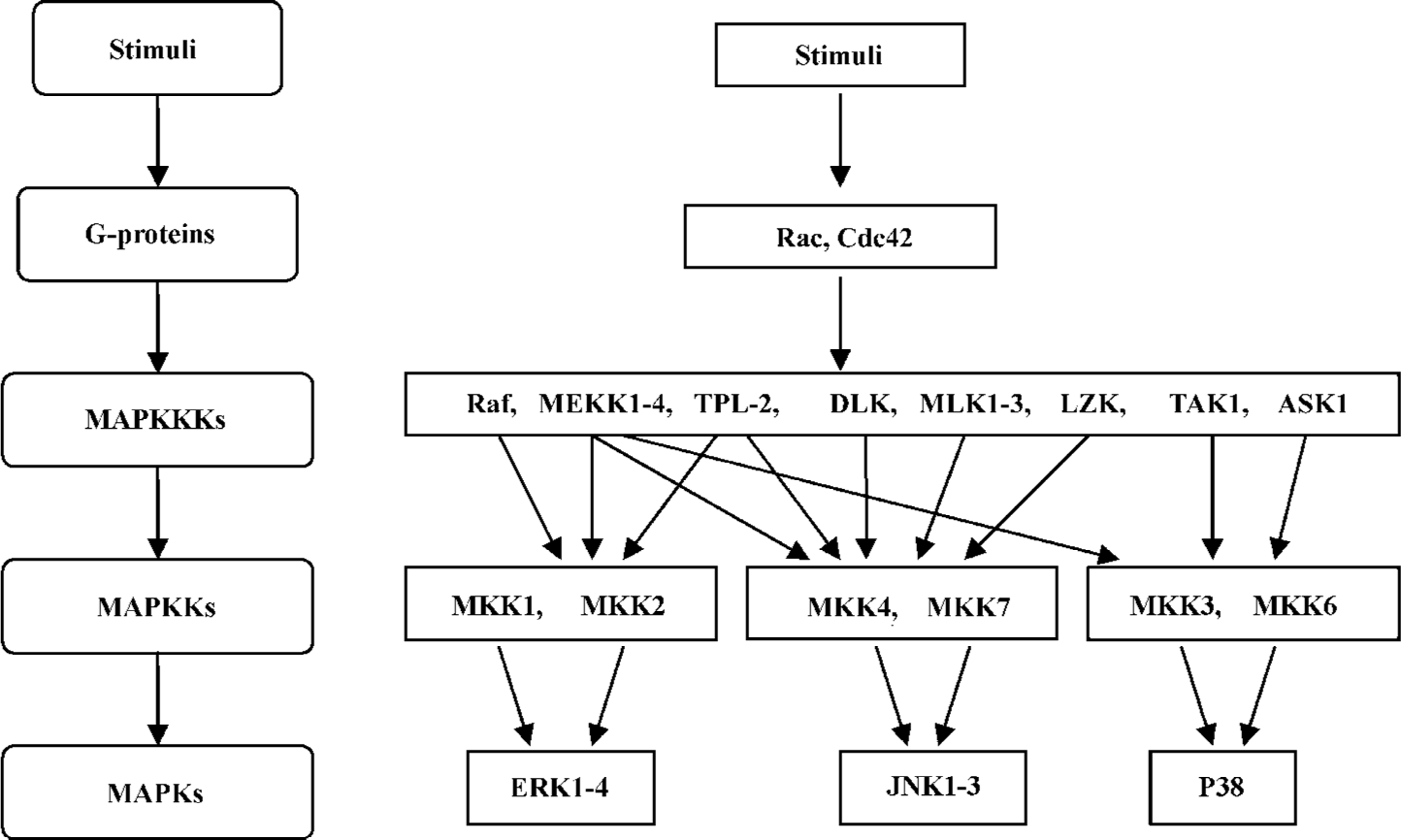
MAPK pathways are conserved in fungi, plants, and mammals. MAPK cascades contain at least 3 protein kinases in series. They are MAPK kinase kinases (MAPKKK, also known as MEKK), MAPK kinases (MAPKK, also known as MEK and MAPK). MAPKKK phosphorylate and activate the downstream MAPKK, which in turn phosphorylate and activate MAPK (Figure 1). These kinases are activated by dual phosphorylation[1–3]. As there are many different extracellular signals in the cellular contexts, MAPKKK show high divergence in structure and gene number compared with MAPKK and MAPK. Typically, MAPKKK are regulated through receptor activation, membrane recruitment of adaptor molecules, activation of small GTP-binding proteins, and phosphorylation. The activation of the cascades may also require additional kinases upstream of this 3-tier kinase, as well as scaffold proteins that are able to manage the complexity and specificity of the pathway. The scaffold proteins can bind, organize, and facilitate specific interactions among the components so that specific stimuli can produce specific MAPK signaling responses (Figure 2)[2,4]. Because of the implication of MAPK in developmental processes and the survival/death responses of cells to their environment, the MAPK pathway has become the focus of attention in recent years[5].
In mammals, there are 3 subfamilies of MAPK that have been identified up to now: c-Jun N-terminal protein kinases (JNK)/stress-activated protein kinases (SAPK), p38 MAPK, and ERK (Figure 1)[2,6]. Following the same signaling arrangement, each individual MAPK pathway responds to specific stimuli and then regulates their specific substrates. This review will focus on the JNK MAPK pathway and its involvement in a variety of diseases.
JNK MAPK pathway
The mammalian JNK were initially described as SAPK because they are activated in response to a variety of environmental stresses[7,8]. The JNK pathway is also recognized to respond to cytokines (eg, TNF and interleukin-1) and growth factors[9].
JNK is a multifunctional kinase involved in many physiological and pathological processes. The JNK pathway plays a major role in apoptosis in a variety of death paradigms. For example, the JNK pathway is required for neuronal cell death induced by many apoptotic stimuli, including nerve growth factor (NGF) deprivation, trophic support withdrawal, DNA damage, oxidative stress, α-amyloid exposure, low potassium, excitotoxic stress, 6-OHDA, UV irradiation, and tumor necrosis factor[10–19].
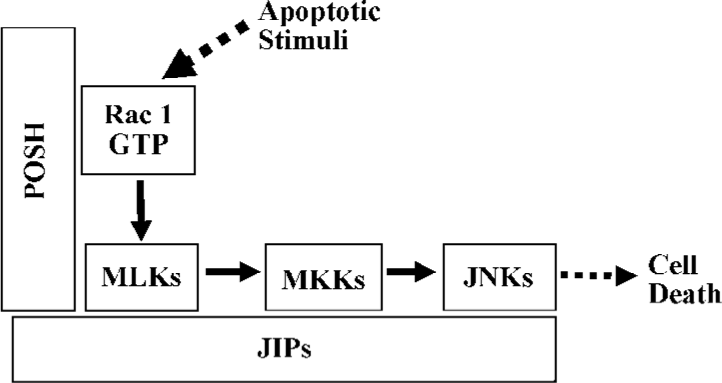
The JNK pathway cascade involved in neuronal cell death has been widely studied. It mainly contains activated Rac1/Cdc42, mixed-lineage kinases (MLK) and/or ASK1, MKK 4 and MKK7, and JNK in series[1,10,15–18,20]. In addition, other components are involved in the activation of the JNK pathway[16,18,19]. Furthermore, plenty of SH3 (POSH) and JNK interacting proteins (JIP) have been shown to act as a scaffold protein to form a multiprotein complex with other JNK apoptotic cascade elements[15,21,22].
In mammals, there are 3 JNK genes: Jnk1, Jnk2, and Jnk3 on 3 different chromosomes, and each mammalian JNK gene has alternative splicing forms so there are at least 10 different JNK proteins of 46–55 kDa identified[23]. JNK1 and JNK2 are ubiquitously expressed, but the expression of JNK3 is mainly restricted to central nervous system (CNS) neurons (high level), cardiac smooth muscle, and testis (low levels)[10]. There is a different substrate affinity among these 10 isoforms[23]; therefore, different JNK isoforms could play specific roles in multicellular organisms, and there is also complementation between the JNK genes.
As to the substrates of JNK, initially studies found that JNK can interact with and phosphorylate the N-terminal transactivation domain of c-Jun and thereby enhance its ability to transactivate related gene expression, so it is called JNK[24]. However, ongoing studies show JNK can phosphorylate a variety of substrates, including additional transcription factors and even some non-nuclear proteins. In addition to c-Jun, JNK can phosphorylate transcription factors such as JunB, JunD, c-Fos, and ATF. These transcription factors, together with c-Jun, constitute activator protein-1 transcription factors, which can regulate the expression of several stress-responsive genes[25]. There are other transcription factors such as ElK1, NFAT, and p53 that can also be phosphorylated by JNK[28]. JNK mediates apoptosis not only through its effects on gene transcription, but also through a transcriptional-independent mechanism[29]. Several studies have demonstrated that JNK is able to phosphorylate both pro- and anti-apoptotic proteins and regulate their activity. For example, JNK can phosphorylate Bcl-2 and Bcl-xL and diminish their anti-apoptotic activity[30–32]. JNK can also phosphorylate the pro-apoptotic protein Bim and Bmf to promote their pro-apoptotic effects[33,34].
Involvement of the JNK pathway in diseases
The JNK pathway is involved in many physiological processes such as embryonic morphogenesis and naturally-occurring programmed cell death. The specific roles that JNK plays depend on the cellular context[10]. Under pathological conditions, the unusual activated JNK pathway can cause pathological cell death and different diseases.
JNK pathway and neurological disorders Mounting evidence suggests that the JNK pathway plays critical roles in the pathogenesis of many neurological disorders, including ischemic stroke, Parkinson’s disease (PD), polyglutamine diseases, amyotrophic lateral sclerosis, auditory hair cell degeneration in addition to different tauopathies, which include Alzheimer’s disease (AD), Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease, familial frontotemporal dementia, and parkinsonism.
Hyperphosphorylation and the accumulation of tau in neurons (and glial cells) is one of the main pathological hallmarks in AD. Increased levels of activated JNK have been found in brain homogenates in all the tauopathies. Strong active JNK immunoreactivity has been observed to be restricted to neurons and glial cells containing hyperphos-phorylated tau, as well as in dystrophic neurites of senile plaques in AD[35]. The accumulation of amyloid beta peptide (Abeta) is believed to be an early and critical event leading to synapse and neuronal cell loss in AD. Abeta itself is toxic to neurons in vitro, and the expression of Abeta in vivo causes the loss of synapses and neurons in the brain in animal models[36]. Interestingly, the increased expression level of activated forms of JNK and p38 and hyperphos-phorylated tau containing neurites has been observed in mice transgenic of human amyloid precursor protein-695 with the Swedish familial AD mutations (Tg2576) and a P264L familial AD mutation introduced by targeting the presenilin-1 gene[36]. In the Tg2576 model, JNK activity is associated with the 2 hallmarks of AD: the amyloid deposit and the hyper-phosphorylated Tau protein[36].
Reactive oxygen species (ROS) has been considered to be the major cause of Abeta toxicity and it is believed that the Abeta-triggered generation of ROS leads to the activation of the JNK pathway, which in turn phosphorylates tau in neurites surrounding amyloid deposits[35,37]. Corresponding evidence has been reported from the autopsies of AD patients who participated in the amyloid-beta immunization trial and died from the immunization-induced encephalitis. The activation of JNK was reduced together with decreased tau hyperphosphorylation of aberrant neurites in association with decreased amyloid plaques in both Tg2576 mice and human brains[35]. Thus, pharmacological inhibitors of JNK may also offer neuroprotection for AD.
JNK signaling has been implicated in the animal model of MPTP-induced degeneration of neurons in the substantia nigra. Following MPTP injection, the obvious activation of JNK, the downstream substrate of JNK, c-Jun, and the upstream kinase MKK4 have been detected in the substantia nigra, correlating with the death of dopaminergic neu-rons[12,38]. A peptide JNK inhibitor interferes with the interaction between JNK and JIP, and inhibits MPTP-induced JNK activation and cell death in the substantia nigra[39]. Gene targeting studies have shown that JNK2 and JNK3 play a vital role in MPTP-induced neuronal death in the substantia nigra. These results, along with the neuroprotective effects by the JNK-pathway inhibitor CEP-1347 in animal MPTP models, have propelled JNK signaling to be a promising target for the pharmacological treatment of the PD[40].
For ischemic stroke, increased activity of JNK was found to co-localize with TUNEL-labeling in the peripheral area of focal ischemia animal models[41]. JNK3 knockout mice are remarkably resistant to kainic acid-induced excitotoxicity[10]. Subsequent studies further showed that JNK3-deficient mice had increased resistance to a global ischemia-hypoxia model of stroke[42]. JNK3 deficiency renders reduced Bim and Fas expression after stroke, and JNK3-null hippocampal neurons have less cytochrome c release following oxygen-glucose deprivation[42]. Furthermore, mice lacking the JNK signaling scaffold protein JIP1 have increased resistance to the gluta-mate excitotoxicity[43] and reduced infarct size in a focal ischemia model of stroke[44]. Knockdown of another scaffold protein POSH is neuroprotective following cerebral ischemia in the rat hippocampus[45]. Taken together, these studies indicate that the JNK pathway may play an important role in ischemic cell death. It offers an opportunity to develop or apply inhibitors of JNK to treat neuronal cell death incurred by ischemia.
JNK pathway and other diseases It has been recognized that the JNK pathway serves as a major “on” switch of programmed cell death in response to a variety of stimuli[46]. In addition to the well-established roles of JNK in neurological disorders, the JNK signaling pathway plays unexpected pathological roles in other diseases, including type 2 diabetes, cancer, stroke, heart disease, and inflammatory diseases.
Type 2 diabetes is the most prevalent and serious metabolic disease hallmarked by pancreatic beta-cell dysfunction and insulin resistance. Under diabetic conditions, oxida-tive stress and endoplasmic reticulum stress are induced in various tissues, leading to the activation of the JNK pathway[47].
Recent studies have demonstrated that JNK plays a central role in modulating insulin action and the pathogenesis of obesity, fatty liver disease, and type 2 diabetes[48]. JNK can directly phosphorylate IRS-1 at several sites, including Ser307[49]. There is increased JNK-dependent IRS-1 Ser phosphorylation in obesity that leads to reduced insulin-stimulated IRS-1 Tyr phosphorylation. In obese animals, knockout of the JNK-1 gene, or the expression of a dominant-negative JNK isoform, results in the reversal of obesity-induced IRS-1 Ser phosphorylation and substantial protection against insulin resistance and defective insulin receptor signaling[4,48], while the exogenous expression of JNK in the adult liver results in severe insulin resistance in mice[4]. JIP1 in knockout mice creates a phenotype very similar to JNK-1 deficiency with reduced JNK activity and increased insulin sensitivity[50]. Meanwhile, a point mutation in JIP1 has been identified in type 2 diabetes patients, providing crucial genetic evidence for the role of JNK and JIP1 in the pathogenesis of type 2 diabetes in humans[51].
It is likely that JNK activity modulates islet function and/or survival in different ways. First, JNK is involved in islet cell inflammation and death mediated by cytokines[52–54]. Second, JNK activation might cause β-cell dysfunction and defective insulin production, thereby contributing to the development of frank diabetes[55]. Third, administration of SP600125, an inhibitor of JNK, improves glucose-stimulated insulin production in isolated islets in the db/db model of obesity and diabetes[56]. Therefore, JNK might integrate defects in insulin secretion with peripheral insulin resistance in type 2 diabetes through its actions in pancreatic β-cells as well as peripheral sites of insulin action.
Taken together, there is very strong evidence that abnormal JNK activation is a critical event in the deterioration of glucose homeostasis and suppression of JNK in diabetic mice. Thus, the JNK pathway plays a central role in the pathogenesis of type 2 diabetes and could be a potential target for diabetes therapy.
Development of JNK pathway inhibitors
In light of the mounting evidence indicating the involvement of JNK signaling in different diseases, it is not surprising that JNK signaling has been of particular interest for drug development.
The complete elimination of JNK genes either alone or in combination has been the initial approach in understanding JNK function. It is very helpful for finding out the specific JNK target for diseases. For example, JNK3 is primarily localized in CNS neurons. Studies from JNK3 knockout mice indicate that JNK3 is an attractive target for neurodegenera-tion therapy in which neuronal cell death should be prevented[10]. In addition, JNK1 knockout can protect mice from obesity-induced insulin resistance, so JNK1 provides a target for the treatment of type 2 diabetes[48]. However, the “knockout” strategy can not be used in disease therapy because the long-term and complete loss of JNK pathway would cause unwanted side-effects. So many alternative approaches that can partly and temporally inhibit JNK function have been developed in recent years. These approaches include antisense techniques, RNAi techniques, and chemical and peptide inhibitors. Some commonly used inhibitors proved to be efficient for use both in vitro and in vivo, indicating that the JNK pathway is suitable for therapeutic use.
There are mainly 3 types of inhibitors thoroughly studied up to now. The first is the ATP-competitive inhibitor of the JNK pathway such as CEP-1347 and SP600125. Most of them are small organic compounds. Most drug discovery programs have focused on designing this kind of inhibitors. They occupy the ATP-binding site of the protein kinase, which is structurally similar in all kinases, so the phosphorylation of substrates is blocked. The ATP-competitive inhibition can be specific, with each inhibitor recognizing the specific ATP-binding site of each kinase. Up to now, at least 40 structurally-different small molecules have been described[40]. However, there is also the disadvantage that the efficacy of the ATP-competitive inhibitor can be reduced by the high endogenous levels of ATP. This has been observed in SP600125[57]. In addition to the ATP-binding site, other sites on the kinase can also provide the target for inhibition, so the second kind of inhibitor targets the substrate-binding site. Based on this mechanism, many peptide inhibitors and dominant-negative mutants against kinases have been designed. The non-phosphorylatable substrate analogue provides a good example of this kind. The third kind of inhibitor targets the allosteric regulatory sites. Either peptides or chemical non-peptides can be used if they can bind to the regulatory sites and block the phosphorylation.
CEP-1347 CEP-1347, originally named KT7515 or 3,9 bis-[(ethylthio)methyl]-K252a, was initially identified as a derivative of the natural compound K252a. The indolocar-bazole K252a has both survival-promoting and neurotrophic effects. Compared to K252a, CEP-1347 is more suitable for therapeutic use because it retains the desirable properties of K252a and minimizes the undesirable effects[58]. For example, the natural compound K252a can inhibit the high affinity nerve growth factor receptor Trk, protein kinase C, myosin light chain kinase and cAMP-dependent protein kinase[58], and calmodulin-activated enzymes[59]. These inhibitory activities greatly limit its use as a neuronal therapeutic agent. CEP-1347 has much lower inhibitory activity towards these kinases.
Studies have shown that CEP-1347 can serve as a neurotrophic molecule to maintain the trophic status of neurons[60]. In addition, CEP-1347 can inhibit the activation of the JNK pathway and thereby prevent neuronal death in both cell culture and animal models. Pretreatment with CEP-1347 prevents β-amyloid-, NGF-withdrawal-, oxidative-stress-, and UV irradiation-induced cell death in PC12 cells and rat sympathetic neurons[11,13]. In the MPTP-model of PD, CEP-1347 can suppress JNK and MKK4 phosphorylation, as well as MPTP-induced cyclooxygenase-2 expression, and protects dopaminergic neurons[61,62]. In another model of PD-intrastriatal injection of 6-hydroxydopamine in rats, the CEP-1347 analogue CEP-11004 also diminishes the apoptotic death of dopaminergic neurons[40]. Taken together, these studies strongly implicate the protective functions of CEP-1347 in animal models of PD.
In addition to PD models, the subcutaneously delivery of CEP-1347 attenuates noise-induced hearing loss and prevents neomycin and aminoglycoside-induced hair cell death in guinea pigs[63,64]. CEP-1347 has also been applied to other disease models that result from JNK activation such as pancreatitis[65] and pulmonary fibrosis[66].
As to the mechanism of inhibition, Maroney et al found that the direct biochemical target of CEP-1347 was the MLK family, the upstream kinase activator of JNK in the JNK pathway[67]. By reducing the kinase activity of MLK, CEP-1347 selectively inactivates the JNK signaling pathway, but spares the ERK and p38 MAPK pathways[60].
CEP-1347 has undergone phase II/III clinical trials for neuroprotection in PD. It has been discontinued for lack of clinical efficacy. Although disappointing, we have to keep in mind that the dose of CEP-1347 used in the trials was not high enough in order to avoid potential adverse effects. In addition, although CEP-1347 can be effective in preventing neuronal cell death, it may not improve the symptoms since dopaminergic neurons were lost before treatment in PD patients.
SP600125 The structure of SP600125 (anthrax[1,9-cd][yrazol-6{2H}-one]) is consistent with other known kinase inhibitors. SP600125 is a reversible ATP-competitive inhibitor of protein kinases. It targets all 3 JNK gene products (JNK1, JNK2, and JNK3) by competing with the ATP-binding site[57]. Since it inhibits JNK MAPK rather than upstream components of the pathway, SP600125 is likely to produce much more specificity of therapy effects than CEP-1347. SP600125 has been shown to be effective in suppressing cell death in the presence of different apoptotic stimuli[18]. In the MPTP model of PD, SP600125 can partially restore dopamine levels[68]. SP600125 has also been demonstrated in animal models of asthma[69]. However, there are also some disadvantages regarding this JNK inhibitor. First, it inhibits all JNK isoforms. The JNK gene knockout studies mentioned earlier have revealed isoform specific roles of JNK, so compounds showing isoform selectivity would be preferred for specific disease therapy. Second, at higher concentrations, SP600125 has more effects than expected since MKK3 and MKK6, and MKK4 and MKK7 are also inhibited[57]. As MKK3 and MKK6 are upstream activators of the p38 MAP kinase signaling pathway, the effects of SP600125 may not be entirely due to the inhibition of JNK signaling especially at higher concentrations. So we have to test the specificity of SP600125 towards other kinases to evaluate its therapeutic use. Third, because JNK is a terminal kinase in the cascade, the inhibition of JNK may produce more general effects compared to upstream kinase inhibition. Finally, SP600125 has very poor water solubility. That is why its application in neurological disorders and other disease models is limited so far.
AS601245 AS601245 (1,3-benzothiazol-2-yl (2-[[2-(3-pyridinyl)ethyl] amino]-4 pyrimidinyl) acetonitrile) has been shown to inhibit the JNK signaling pathway similar to SP600125. It inhibits JNK3 with IC50 of 70 nmol/L and IC50 for JNK1 and JNK2 at 150 and 220 nmol/L, respectively[70]. Thus, AS601245 appears to be a more selective inhibitor of JNK3 at a low dose. AS601245 provides significant protection against the delayed loss of hippocampal CA1 neurons in a gerbil model of transient global ischemia. A significant neuroprotec-tive effect of AS601245 was also observed in rats after focal cerebral ischemia[70,71]. AS601245 has also been shown to decrease cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats[72]. The in vitro and in vivo anti-inflammatory potential of AS601245 was investigated recently and found to demonstrate efficacy in an experimental model of rheumatoid arthritis[73]. It would be intriguing to investigate whether AS601245 has cytoprotective effects in animal models of neurodegen-erative diseases.
Peptide inhibitors of the JNK pathway Peptide inhibitors target the protein/peptide substrate binding sites or regulatory sites of the protein kinases. This provides a more specific inhibition mechanism than ATP-competitive inhibitor. Many non-phosphorylatable substrate analogues have been designed on the basis of substrate-competition.
As to the regulatory site inhibition, a non-substrate JIP has been found in a 2-hybrid screen[21]. As mentioned earlier, JIP act as scaffold proteins to form a multiprotein complex with other JNK apoptotic cascade elements. However, JIP1 overexpression can block JNK activity in mammalian cells[21] and cell death in several physiological models[74]. This is probably because excess scaffold results from overexpres-sion compared to the limited amounts of other JNK pathway elements that disrupt the integrity of each complex.
The D-JNKI-1 peptide, a cell penetrating JNK inhibitor, was constructed by linking the 20 amino acid segment of a JNK-binding domain (JBD) from JIP1 to a 10 amino acid HIV-TAT transporter sequence and by synthesizing a highly protease resistant form of the JBD that doubles its intracellular half-life[53]. It has been demonstrated to prevent loss of hearing following either electrode insertion trauma or loss of both hearing and hair cells following exposure to ototoxic levels of neomycin[75]. D-JNKI also showed a remarkable neuro-protective effect in both the transient and permanent middle cerebral artery occlusion (MCAO) model of stroke[76]. The protection correlated with the prevention of an increase in c-Jun activation and c-Fos transcription. Importantly, DJNKI reduces the infarct volume, even when administered at 6–12 h after transient MCAO. In view of its potency and long therapeutic window, this protease-resistant peptide is a promising neuroprotective agent for stroke.
Conclusions and perspectives
It has been recognized that the JNK pathway plays important roles in many physiological and pathological conditions. Therefore, JNK pathway components represent potential therapeutic targets for the treatment of related diseases, including neurodegenerative diseases, cancer, diabetes, hearing loss, heart diseases, inflammatory diseases, and autoimmune diseases. Several kinds of inhibitors have been developed, targeting different levels of the JNK signaling cascade and some of them, such as CEP-1347 and SP600125, have been used in clinical trials. Although there are still some problems limiting the use of these inhibitors such as the substrate specificity, the crosstalk between the JNK and other signaling pathway, and potential side effects, including the possibility to induce tumor formation, we are hopeful that effective drugs targeting the JNK pathway will be developed for relevant diseases in the near future. To reach this goal, further efforts are required to learn more about the physiological roles of the JNK pathway components while attention should be paid to develop more specific inhibitors. In addition, long-term clinical studies are needed to observe whether JNK pathway inhibitors would lead to tumor formation and tumor metastasis in some circumstances.
References
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239-5.
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev 2002;12:14-21.
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001;22:153-83.
- Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka TA, et al. Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem 2004;279:45803-9.
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov 2003;2:554-65.
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807-69.
- Kyriakis JM, Avruch J. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem 1990;265:17355-63.
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, et al. The stress-activated protein kinase subfamily of c-Jun kinase. Nature 1994;369:156-60.
- Ichijo H. From receptors to stress-activated MAP kinase. Oncogene 1999;18:6087-93.
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997;389:865-70.
- Maroney AC, Finn JP, Bozyczko-Coyne D, O’Kane TM, Neff NT, Tolkovsky AM, et al. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem 1999;73:1901-12.
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, et al. Beta-amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem 2001;77:157-64.
- Trotter L, Panton W, Hajimohamadreza I, Petalidis L, Ward R, Fleming Y, et al. Mitogen-activated protein kinase kinase 7 is activated during low potassium-induced apoptosis in rat cerebellar granule neurons. Neurosci Lett 2002;320:29-32.
- Xu Z, Kukekov NV, Greene LA. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 2003;15:252-61.
- Xu Z, Kukekov NV, Greene LA. Regulation of apoptotic c-Jun N-terminal kinase signaling by a stabilization-based feed-forward loop. Mol Cell Biol 2005;25:9949-59.
- Xu Z, Sproul A, Wang W, Kukekov N, Greene LA. Siah1 interacts with the scaffold protein POSH to promote JNK activation and apoptosis. J Biol Chem 2006;281:303-12.
- Xu Z, Greene LA. Activation of the apoptotic JNK pathway through the Rac1-binding scaffold protein POSH. Methods Enzymol 2006;406:479-89.
- Wilhelm M, Xu Z, Kukekov NV, Gire S, Greene LA. Proapoptotic Nix activates the JNK pathway by interacting with POSH and mediates death in a Parkinson disease model. J Biol Chem 2006. [Epub ahead of print].
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol 2001.4713-24.
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, et al. A cytoplasmic inhibitor of the JNK signaling transduction pathway. Science 1997;277:693-6.
- Kukekov NV, Xu Z, Greene LA. Direct interaction of the molecular scaffolds POSH and JIP is required for apoptotic activation of JNKs. J Biol Chem 2006;281:15517-24.
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 1996;15:2760-70.
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994;76:1025-37.
- Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995;267:389-93.
- Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, et al. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 1998;9:575-85.
- Chow CW, Rico MN, Cavanagh J, Dickens M, Davis RJ. Nuclear accumulation of NFAT4 opposed by the JNK signaling pathway. Science 1997;278:1638-41.
- Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, Henderson S, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcription activities in response to stress. Mol Cell Biol 2001;21:2743-54.
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000;288:870-4.
- Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem 1997;272:25238-42.
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol cell Biol 1999;19:8469-78.
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem 2000;275:322-7.
- Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 2002;277:40944-9.
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl-2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 2003;100:2432-7.
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, et al. Current advances on different kinases involved in tau phos-phorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 2005;2:3-18.
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW, et al. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 2002;22:3376-85.
- Smith WW, Gorospe M, Kusiak JW. Signaling mechanisms underlying Abeta toxicity: potential therapeutic targets for Alzheimer’s disease. CNS Neurol Disord Drug Targets 2006;5:355-61.
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 2004;101:665-70.
- Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA 2001;98:10433-8.
- Kuan CY, Burke RE. Targeting the JNK signaling pathway for stroke and Parkinson’s diseases therapy. Curr Drug Targets CNS Neurol Disord 2005;4:63-7.
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, et al. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci 1998;18:5124-35.
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci USA 2003;100:15184-9.
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev 2001;15:2421-32.
- Im JY, Lee KW, Kim MH, Lee SH, Ha HY, Cho IH, et al. Repression of phospho-JNK and infarct volume in ischemic brain of JIP1-deficient mice. J Neurosci Res 2003;74:326-32.
- Zhang QG, Wang RM, Yin XH, Pan J, Xu TL, Zhang GY. Knock-down of POSH expression is neuroprotective through down-regulating activation of the MLK3–MKK4–JNK pathway following cerebral ischaemia in the rat hippocampal CA1 subfield. J Neurochem 2005;95:784-95.
- Chen YR, Meyer CF, Tan TH. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in radiation-induced apoptosis. J Biol Chem 1996;27:631-4.
- Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, et al. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. Int J Biochem Cell Biol 2006;38:782-93.
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. Hotanisligil, a central role for JNK in obesity and insulin resistance. Nature 2002;420:333-6.
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 2000;275:9047-54.
- Jaeschke A, Czech MP, Davis RJ. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev 2004;18:1976-80.
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, et al. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 2000;24:291-5.
- Ammendrup A, Maillard A, Nielsen K, Aabenhus Andersen N, Serup P, Dragsbaek Madsen O, et al. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000;49:1468-76.
- Bonny A, Oberson S, Negri C, Sauser X, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 2001;50:77-82.
- Abdelli S, Ansite J, Roduit R, Borsello T, Matsumoto I, Sawada T, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 2004;53:2815-23.
- Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 2002;277:30010-8.
- Bennett BL, Satoh Y, Lewis AJ. JNK: a new therapeutic target for diabetes. Curr Opin Pharmacol 2003;3:420-5.
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 2001;98:13681-6.
- Kaneko M, Saito Y, Saito H, Matsumoto T, Matsuda Y, Vaught JL, et al. Neurotrophic 3,9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J Med Chem 1997;40:1863-9.
- Kase H, Iwahashi K, Matsuda Y. K-252a, a potent inhibitor of protein kinase C from microbial origin. J Antibiot (Tokyo) 1986;39:1059-65.
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Murphy CA, et al. Motoneuron apoptosis is blocked by CEP-1347 (KT7515), a novel inhibitor of the JNK signaling path- way. J Neurosci 1998;18:104-11.
- Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J Pharmacol Exp Ther 1999;288:421-7.
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci USA 2003;100:5473.
- Pirvola U, Liang XQ, Virkkala J, Saarma M, Murakata C, Camoratto AM, et al. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci 2000;20:43-50.
- Ylikoski J, Liang XQ, Virkkala J, Pirvola U. Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hear Res 2002;166:33-43.
- Wagner AC, Mazzucchelli L, Miller M, Camoratto AM, Goke B. CEP-1347 inhibits caerulein-induced rat pancreatic JNK activation and ameliorates caerulein pancreatitis. Am J Physiol Gastrointest Liver Physiol 2000;278:G165-72.
- Hashimoto S, Gon Y, Takeshita I, Maruoka S, Horie T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase-dependent pathway. J Allergy Clin Immunol 2001;107:1001-8.
- Maroney AC, Finn JP, Connors TJ, Ddurkin JT, Angeles T, Gessner G, . CEP-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem 2001; 276: 25 302–8.
- Wang W, Shi L, Xie L, Ma C, Li W, Su X, et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res 2004;48:195-202.
- Chialda L, Zhang M, Brune K, Pahl A. Inhibitors of mitogen-activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophilia. Respir Res 2005;15:6-36.
- Carboni S, Hiver A, Szyndralewiez C, Gaillard P, Gotteland JP, Vitte PA. AS601245 (1,3-benzothiazol-2-yl (2-[[2-(3-pyridinyl) ethyl] amino]-4 pyrimidinyl) acetonitrile): a c-Jun NH2-terminal protein kinase inhibitor with neuroprotective properties. J Pharmacol Exp Ther 2004;301:25-32.
- Carboni S, Antonsson B, Gaillard P, Gotteland JP, Gillon JY, Vitte PA. Control of death receptor and mitochondrial-dependent apoptosis by c-Jun N-terminal kinase in hippocampal CA1 neurons following global transient ischaemia. J Neurochem 2005;92:1054-60.
- Ferrandi C, Ballerio R, Gaillard P, Giachetti C, Carboni S, Vitte PA, et al. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br J Pharmacol 2004;142:953.
- Gaillard P, Jeanclaude-Etter I, Ardissone V, Arkinstall S, Cambet Y, Camps M, et al. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J Med Chem 2005;48:4596-607.
- Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem 2002; 277: 10 987–97.
- Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci 2003;23:8596-607.
- Hirt L, Badaut J, Thevenet J, Granziera C, Regli L, Maurer F, et al. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004;35:1738-43.