
High-throughput screening for monoamine oxidase-A and monoamine oxidase-B inhibitors using one-step fluorescence assay1
Introduction
Monoamine oxidase (MAO) is responsible for oxidative deamination of endogenous and xenobiotic amines located at the outer membrane of mitochondria in neuron and non-neuronal cells. Two isoforms of MAO have been identified and designated as MAO-A and MAO-B[1], coded by similar but distinct genes, having different substrate preference and inhibitor specificity[2]. Experimentally, MAO-A preferentially oxidizes serotonin (5-hydroxytryptamine, 5HT), noradrenaline and adrenaline, and is inhibited by low concentrations of clorgyline. In contrast, MAO-B preferentially oxidizes dopamine, b-phenylethylamine (PEA), benzylamine and is inhibited by low concentrations of deprenyl or pargyline[3].
Changes of MAO activity are involved in some central and peripheral nervous system diseases. Extra high MAO-B activity in the brain appears in neurological degenerations involving Alzheimer’s disease, Huntington’s disease, some forms of Parkinson’s disease and normal aging[4]. Abnormal MAO-A activity is implicated in depression, anxiety and psychiatric disorders[5]. Clinically, MAO inhibitors have been found to alleviate symptoms or slow deterioration of these diseases, which impels the necessity of discovering more MAO inhibitors (MAO-I).
The establishment of high-throughput screening (HTS) assays is an essential element in the drug discovery processes. Based on the general reaction RCH2NR1R2+ H2O+O2 —→ RCHO + NHR1R2 + H2O2, available assays to measure MAO activity can be summarized either by determining the rate of product formation or substrates depleted in an MAO-catalyzed reaction: (1) the direct measurement of oxygen consumption by polarographic detection[6]; (2) the detection of oxidized monoamine products by spectrophotometry or radiometric assay[7,8]; (3) liquid chromatography with tandem mass spectrometry measuring MAO activity has been recently applied to HTS, but it has less specificity and limited convenience[9]; (4) the assessment of co-product hydrogen peroxide directly[10] or indirectly, a continuous fluorescence assay with a sensitive, stable H2O2 probe, N-acetyl-3,7-dihydroxyphenoxazine (Amplex Red). Because of its convenience and continuity, this one-step method was considered more suitable to detect the activity of enzymes whose co-product is hydrogen peroxide. However, there has been no establishment detecting MAO-A and MAO-B activity by this method until now. HTS protocol discovering MAO-A-inhibitor (MAO-A-I) and MAO-B-inhibitor (MAO-B-I) applying this assay has not yet been developed.
In the current experiment, we detected both MAO-A and MAO-B activity by this fluorescence probe based method with evaluations of its sensitivity, stability and specificity. The reliability of this assay was valued by comparing MAO-A and MAO-B dynamic parameters with those obtained by traditional assays. In regard to enzyme source facilitations, we purified rat brain mitochondria by graded extractions to obtain higher specific activity. Reaction conditions including enzyme sources, substrates concentrations, incubation volume and reaction time in 384-well format were optimized to make MAO-A-I and MAO-B-I screening protocols with convenience, sensitivity, and low consumption. Moreover, precision parameters were calculated to ensure the confidence of a developed system, intending to provide convenient and robust protocols in MAO-A-I and MAO-B-I screening procedures.
Materials and methods
Materials and animals Horseradish peroxidase (HRP), hydrogen peroxide, benzylamine, serotonin, Triton X-100, bovine serum albumin, Cu-Zn superoxide dismutase (Cu-Zn SOD), clorgyline, and deprenyl were purchased from Sigma (St Louis, MO, USA). Amplex Red was from Molecular Probes (Eugene, OR, USA). Other high-graded chemical reagents were commercially available.
Adult male Wistar rats (280‒300 g) were obtained from the Chinese Academy of Medical Sciences, Experimental Animal Center.
Enzyme preparations Different enzyme preparation conditions were conducted to obtain a high specific activity. Mitochondria were isolated according to our developed assay with modifications[11]. In brief, crude mitochondria were obtained by centrifuging rat brain homogenates at 2000×g for 10 min in MSETB buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 0.5 mmol/L ethylenediamine tetra-acetate, 10 mmol/L Tris-HCl and 0.2% bovine serum albumin, pH 7.4). Sonicated crude mitochondria were gained by sonicating crude mitochondria suspension at 20 Hz twice for 20 s with an interval of 10 s and further centrifuging at 16 000×g for 10 min. Washed mitochondria were obtained by resuspending the crude mitochondria in 1:40 (w/v) SET buffer (280 mmol/L sucrose, 0.5 mmol/L EDTA and 10 mmol/L Tris-HCl pH 7.4) and further centrifuged at 16 000×g for 8 min. Mitochondria were then subsequently thrice frozen at -20 °C and thawed at 4 °C. Further purifications were conducted by solubilizing mitochondria in 0.1% Triton X-100 to reduce homogenate interferences.
To further assess MAO-A and MAO-B quantifications in each extraction step, Western blotting assay was carried out as previously described. Equivalent amounts of total proteins were separated by PAGE/SDS. The primary antibody concentration for MAO-A is 1:2000 (Santa Cruz Biotechnology, Santa Cruz, USA), MAO-B 1:1000 (Santa Cruz Biotechnology, Santa Cruz, USA), and anti-actin antibody 1:2000 (Santa Cruz Biotechnology, Santa Cruz, USA). Immunoreactive deposits were detected using horseradish peroxidase-conjugated secondary antibodies and enzyme-linked chemiluminescence according to manufacturer抯 instructions (ECL, Pierce, Rockford, IL, USA)
Suspension of each purification step was stored at -40 °C in aliquots until required for experiment within 48 h. Specific activity of MAO-A or MAO-B and inhibition properties of clorgyline to MAO-A or deprenyl to MAO-B were examined by fluorescence assay. Protein quantification was evaluated by Lowry’s assay[12].
MAO activity measurement The experiment was performed by optimization at 37 °C (pH 7.4) in 384-well microplate (Greiner, Bio-One, Germany). The reaction mixture contained total enzyme protein at the final concentration of 0.2 mg/mL, and 0.02 mmol/L serotonin as a substrate for MAO-A, 0.01 mmol/L benzylamine as a substrate for MAO-B, with 10 U/mL HRP, 2.5 μmol/L Amplex Red and 40 U/ml Cu-Zn SOD previously supplemented. Amplex Red was oxidized into resorufin (Figure 1) in the reaction, directly detected at 560±10 nm at excitation and 590±10 nm at emission. As a blank control, an identical vehicle solution for samples was added in place of enzyme solution. The Km value of serotonin to MAO-A and that of benzylamine to MAO-B were determined by adding graded concentrations of each substrate to the reaction system. At the same time, time and concentration dependent manners of the standard solution were obtained by applying series of hydrogen peroxide concentrations in Figure 6C.

Evaluations Different reagent combinations were conducted with and without substrates or enzyme incubated in the reaction system with the fluorescence density demonstrated in Figure 5. Fluorescence in the MAO-A and MAO-B catalyzed reaction at different time points was detected with the time-dependent manner.

HTS protocol HTS procedures were performed on conditions of optimized reaction volume as 50μL, in which contained optimized concentrations of enzyme, substrates, and sample. Incubation time of MAO-A catalyzed reaction was 60 min and that of MAO-B catalyzed reaction was 45 min. In primary screening, the samples were examined on their inhibitory potency on MAO-A and MAO-B activity. During the protocol, clorgyline and deprenyl were positive controls in MAO-A inhibitor screening and MAO-B inhibitor screening, respectively, with PBS buffer as the negative control. In further screening, compounds were serially diluted into graded concentrations and the inhibition was measured with IC50.
Data processing Inhibition of samples on MAO activity was represented by the percentage of enzyme activity relative to that of the negative control. Data processing and validation were undertaken according to our standard laboratory data processing technology. Precision parameters involving coefficient of variation (CV), signal to background value (S/B), and Z' factor were calculated according to Zhang’s method[13].
Results
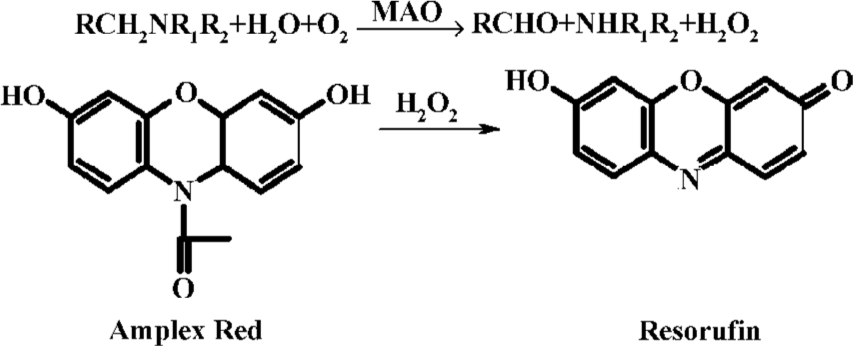
Enzyme preparations Specific activities of MAO-A and MAO-B in different preparation steps are shown in Table 1. Taking intra-day evaluation for instance, MAO-A had its highest activity as 52.91±7.54 nmol·min-1·mg protein-1 in sonicated crude mitochondria, while exhibiting a decreasing tendency in the following steps. In contrast, MAO-B demonstrated an increase of activity in the five preparation steps, reaching its peak of 102.34±11.21 nmol·min-1·mg protein-1 at the Triton X-100 treated step.
Full table
Interestingly, MAO-A and MAO-B immunoblotting evaluations were consistent with performances of specific activity. Inference could be obtained from this coincidence that further extraction increased enzyme content ratio in samples instead of demolishing their structure and activity, inversely confirming enhancements on enzyme specific activity in the purifications (Figure 2).
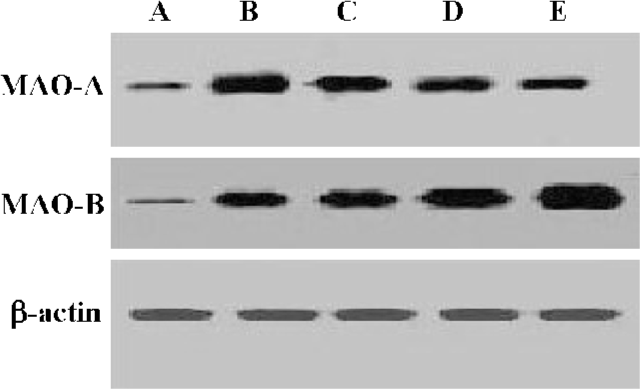
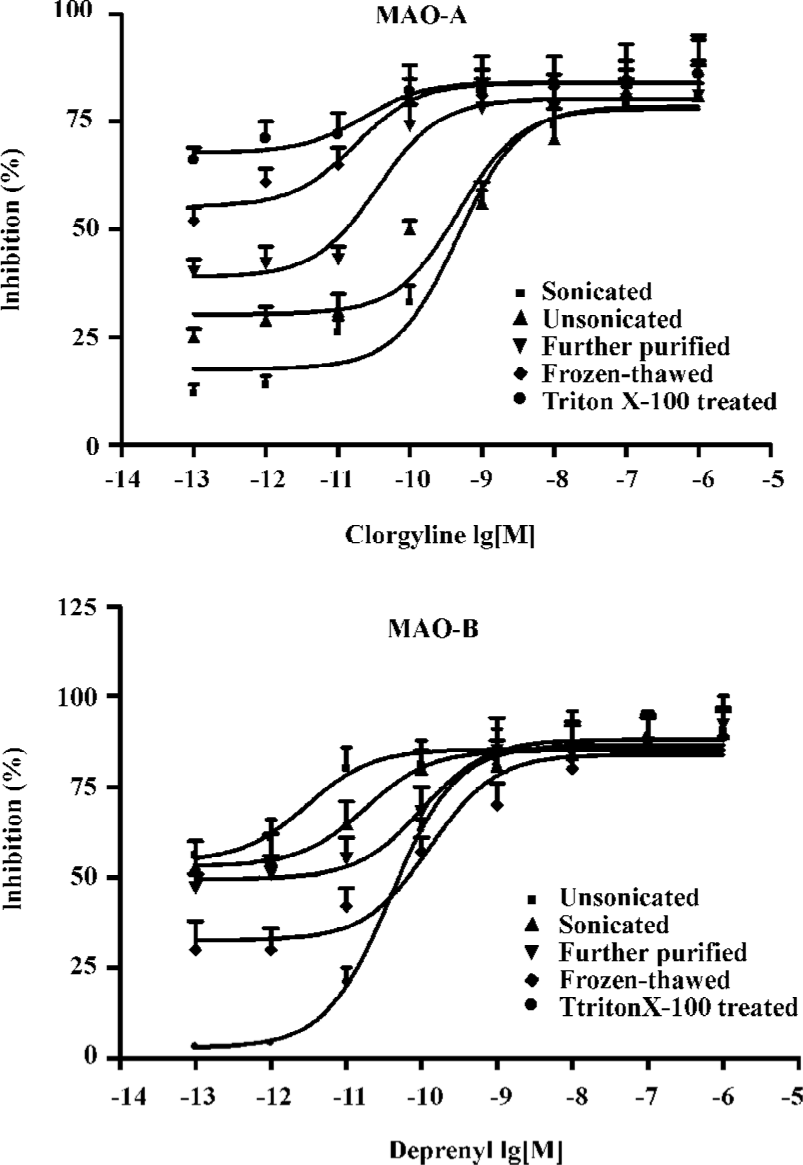
For further evaluation, sensitivities of MAO-A and MAO-B from each step to their specific inhibitors were examined with the tendency curves. MAO-A had the highest sensitivity against clorgyline in the sonicated crude mitochondria step, while demonstrating decreasing tendency in the last three steps. MAO-B demonstrated an increasing sensitivity trend in the five successive enzyme extraction steps. Tendency demonstrated was in accordance with the results shown in Table 1, which demonstrated further different enzyme extraction conditions in MAO-A and MAO-B preparations (Figure 3).
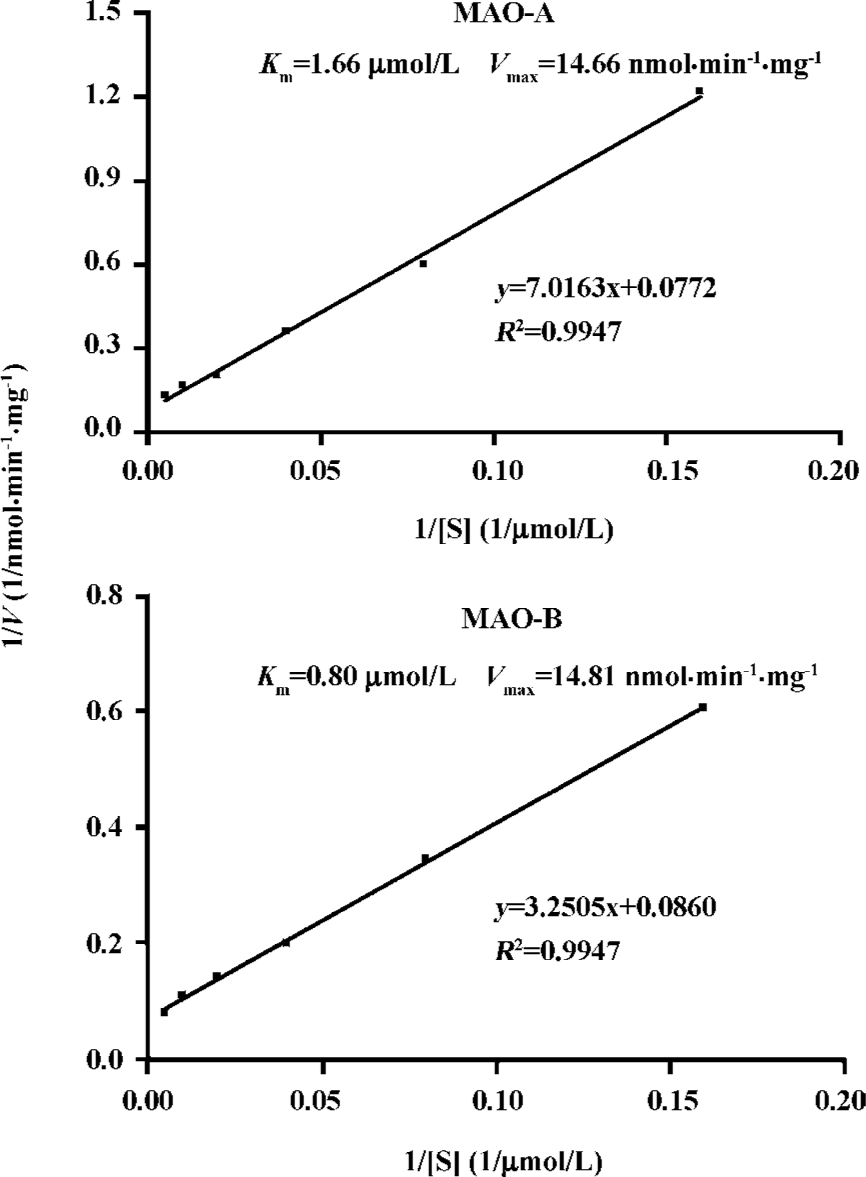
MAO parameters Dynamic parameters of MAO-A and MAO-B from the determined extraction steps were examined with graded concentrations of their substrates added (Figure 4), the Km value of serotonin to MAO-A was 1.66 μmol/L and Vmax was 14.66 nmol·min-1·mg protein-1 in MAO-A catalyzed reaction. In the MAO-B catalyzed reaction, the Km value of benzylamine to MAO-B was 0.80 μmol/L, and Vmax was 14.81 nmol·min-1·mg protein-1.
Evaluations From Figure 5, group VI, which had Amplex Red, HRP, MAO-B, benzylamine co-incubated together in the reaction, demonstrated the highest fluorescence, while group V having Amplex Red, HRP, MAO-A and serotonin together, displayed the second highest fluorescence. How-ever, other groups in the absence of even one reagent demonstrated a low fluorescence, which implied the specificity of this applied assay. Interestingly, group VII, containing Amplex Red, HRP, MAO-A and benzylamine, and group VIII, containing Amplex Red, HRP, MAO-B and serotonin, both had a lower fluorescence also suggesting the sensitivity of this fluorescence assay.
The fluorescence was also detected at successive time points in MAO-catalyzed reaction with graded concentrations of substrates. In Figure 6A, the reaction with 0.02 mmol/L serotonin as its optimum concentration substrate showed a fluorescence peak at 60 min, while in Figure 6B the reaction with 0.01 mmol/L benzylamine as the optimum concentration substrate showed a fluorescence peak at 45 min. In either an MAO-A or MAO-B catalyzed reaction, there existed a stable tendency after the peak in time-dependent curve.
HTS experiment In optimum conditions, compounds in our library were tested for their inhibitory profile on MAO activity, with a final concentration of 10μ/mL in the primary screening process. Results showed that in an MAO-A HTS system 0.6% compounds had relative inhibition higher than 70.1%, and 1.0% compounds had a relative inhibition on MAO-B higher than 70.0%. In further screening processes, there was one compound exerting specific inhibitory potency against MAO-A activity in a concentration-dependent manner, with its IC50 as 0.36μmol/L. Meanwhile, three compounds had specific inhibitory potency against MAO-B activity with their IC50 as 0.13, 0.19, and 0.13μmol/L.
In the present experiment, the applied method was subjected to validation from several batches of quality control samples, with precision presented in Table 1. The inter-day CV was 14.2 and intra-day CV was 5.3 in the applied MAO-A extraction procedure, while in the MAO-B extraction procedure inter-day precision was 10.9 and 4.8 for the intra-day precision. The IC50 value of clorgyline to MAO-A was 2.99 nmol/L, and that to deprenyl was 7.04 nmol/L, in accordance with those detected by the traditional assays[13,14]. The S/B value of the two HTS systems were higher than 3. The Z' factor for MAO-A-I HTS performance was 0.71±0.03 and 0.75±0.03 for the MAO-B-I HTS system.
Discussion
Abnormal MAO changes are involved in neurological diseases, however, most of the available MAO inhibitors have undesirable side effects[16]. This means that more MAO inhibitors need to be discovered. By using robotic automation system to test numbers of compounds against novel biological targets, high-throughput screening (HTS) dramatically accelerates the pace of drug discovery with large amounts of drug candidates efficiently screened out. How-ever, until now, no HTS assay for MAO inhibitors has been developed by appropriately convenient, sensitive and efficient methods.
In retrospect, several conventional methods detecting MAO activity are available but most of them are not suitable for rapid and continuous screen protocols for large amounts of samples. For example, the direct measurement of oxygen consumption needs a very rigid condition control[4]. Other methods such as spectrophotometry or liquid chromatography[17,18] used for analyzing aldehydes formed in the reaction still can not be conveniently utilized in HTS because of low specificity and limited sensitivity. Low sensitivity of an analytical method inevitably requires high concentrations of reaction substances and long incubation periods, resulting in significantly high consumable costs and low throughputs. The radiometric approach using 14C-labeled substrates is more specific and sensitive, but handling of radioactive material or isolation of products by extraction is much more troublesome and costly. Therefore, HTS processes for MAO inhibitors require an MAO detection assay with high specificity, sensitivity, and convenience.
Because MAO acting on its substrates also generates co-product hydrogen peroxide generally independent to the substrate, spectrometric assays designed to directly detect hydrogen peroxide production have been applied to estimate MAO activity or enzyme classification in recent years. However, because direct detection on absorption of hydrogen peroxide is often interfered by crude tissue homogenates at the wavelength of 230 nm, it is not suitable to use HTS protocol with its low sensitivity. Indirect measurement on hydrogen peroxide production based on the HRP-coupled reaction system has only ever been conducted with a hydrogen peroxide-sensitive probe, Amplex Red in human leukocytes[19]. But there have been no studies on the feasibility of detecting fluorescent MAO activity with Amplex Red. HTS protocols applying this assay have not been developed, either.
Attempts to apply this assay to MAO-I HTS platform have been carried out in the current experiment, with sensitivity, specificity and stability evaluations. The low fluorescence background of Amplex Red, homogenates and the individual substrate indicated the high sensitivity of this assay. The fluorescence demonstration in Figure 5 also confirmed specific substrates to their specific MAO subtypes. The sensitivity of the protocol was also proven by the concentration-dependence effect and time-dependence manner shown in Figure 6, in which the peak of MAO-A catalyzed reaction at 60 min and MAO-B catalyzed reaction at 45 min was shown in the present reaction system. Interestingly, it could also be noticed that a stable platform after the reaction peak existed in time-dependence curve, together with time-dependence manner with hydrogen peroxide as the standard solution demonstrating the stability of this applied fluorescence assay.
Considering that the production of hydrogen peroxide was also formed by other oxidative enzymes present in enzyme preparations, Cu-Zn SOD was previously added in the reaction system, preventing the auto-oxidation of Amplex Red that interfered with the quantitative assessment of low rates of hydrogen peroxide production in the MAO-catalyzed reaction. Even with this procedure, the applied assay was still a one-step method with specificity and sensitivity, enabling a convenient platform for this HTS system.
For facilitation, rat brain tissues were chosen as enzyme preparation sources. Regarding the fact that endogenous MAO inhibitors may cause under-determination in the activity evaluation[20], we tried to perform different enzyme extraction protocols to get a high enzyme quality. By optimization, the optimum enzyme preparation conditions were determined by examining the specific activity of enzyme and sensitivity to their specific inhibitors at different extraction steps. As shown in Table 1, MAO-B had its highest activity in the Triton X-100 treated step of the present assay, in which the purification of MAO-B was enhanced or endogenous MAO inhibitors may be washed out, also reversely confirming the existence of endogenous MAO inhibitors. However, MAO-A had very low activity in the last three steps indicating its activity loss in the further extraction procedures, which may not only be a result of its low content in the rat brain but also its comparative vulnerability as outlined in a previous study[21]. From the consistence of MAO-A and MAO-B demonstrations in immunoblotting evaluations with performances of specific activity measurement, inference could be deduced that further extraction increased enzyme content ratio in samples, which confirmed the enhancement of enzyme specific activity in purifications. Figure 3 illustrates the sensitivity of MAO against their inhibitors, in which MAO-A had its highest sensitivity against clorgyline in the sonicated crude mitochondria step, while displaying a decreasing tendency in the last three steps. MAO-B had an increasing sensitivity trend in the five successive extraction steps further confirming the sensitivity of this assay.
Dynamic parameters of MAO-A and MAO-B were measured to evaluate the reliability of this applied assay. Results demonstrated that the Km value of serotonin to MAO-A was 1.66μmol/L, while that of benzylamine to MAO-B was 0.80μmol/L and the IC50 value of clorgyline to MAO-A was 2.99 nmol/L, while that of deprenyl to MAO-B was 7.04 nmol/L. These matched those obtained by the traditional assays[14,15].
During the development of the HTS model, other optimizations were also conducted involving concentrations of reaction substances such as enzyme, substrates, samples and the incubation volume minimization to 50μL with obtained data reproducible. The Z' factors were calculated to evaluate and validate the quality of the overall assay. Results revealed that the Z' factor was both higher than 0.7 in the MAO-A-I and MAO-B-I HTS performances, indicating high quality and reproducibility of the established HTS protocols.
Taken together, the fluorescence probe based and HRP-coupled fluorescence assay in MAO detection was evaluated as sensitive, reliable, and convenient in the present paper. It can not only be used for MAO activity assay and MAO parameters profile but is suitable for HTS processes. The established HTS models applying this one-step fluorescence were robust for discovering MAO-A and MAO-B inhibitors. This was achieved with high efficiency, reproducibility, and low consumption of HTS characterizations.
With the development of recombinant technology, commercial enzymes as recombinant proteins will be applied in this HTS protocol in our future studies for further standardizations and lead-compound evaluations. This applied assay could prospectively be extended to detect other biological compounds or enzymes that also require further investigation.
References
- Abell CW, Kwan SW. Molecular characterization of monoamine oxidases A and B. Prog Nucleic Acid Res Mol Biol 2001;65:129-56.
- Nakagawasai O, Arai Y, Satoh SE, Satoh N, Neda M, Hozumi M, et al. Monoamine oxidase and head-twitch response in mice. Neurotoxicology 2004;25:223-32.
- Sandler M. My fifty years (almost) of monoamine oxidase. Neuro-toxicology 2004;25:5-10.
- Rehman HU, Masson EA. Neuroendocrinology of ageing. Age Ageing 2001;30:279-87.
- Jean CS. Cloning, after cloning, knock-out mice, and physiological functions of MAO A and B. Neurotoxicology 2004;25:21-30.
- Averill-Bates DA, Agostinelli E, Przybytkowski E, Mateescu MA, Mondovi B. Cytotoxicity and kinetic analysis of purified bovine serum amine oxidase in the presence of spermine in Chinese hamster ovary cells. Arch Biochem Biophys 1993;300:75-9.
- Zhou JJ, Zhong B, Silverman RB. Direct continuous fluorometric assay for monoamine oxidase B. Anal Biochem 1996;234:9-12.
- Yoshimi K, Kozuka M, Sakai J, Iizawa T, Shimizu Y, Kaneko I, et al. Novel monoamine oxidase inhibitors, 3-(2-aminoethoxy)-1,2朾enzisoxazole derivatives, and their differential reversibility. Jpn J Pharmacol 2002;88:174-82.
- Yan ZY, Caldwell GW, Zhao BY, Reitz AB. A high-throughput monoamine oxidase inhibition assay using liquid chromatography with tandem mass spectrometry. Rapid Commun. Mass Spectrom 2004;18:834-40.
- Stevanato R, Vianello F, Rogo A. Thermodynamic analysis of the oxidative deamination of polyamines by bovine serum amine oxidase. Arch Biochem Biophys 1995;324:374-8.
- Zhang HX, Du GH, Zhang JT. Ischemic pre-conditioning preserves brain mitochondrial functions during the middle cerebral artery occlusion in rat. Neurol Res 2003;25:471-6.
- Lowry OH, Rosebrougy NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265-71.
- Zhang JH, Chuang TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67-73.
- Saura J, Kettler R, Prada MD, Richards JG. Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J Neurosci 1992;12:1977-99.
- Hirata M, Kagawa S, Yoshimoto M, Ohmomo Y. Synthesis and characterization of radioiodinated MD-230254: a new ligand for potential imaging of monoamine oxidase B activity by single photon emission computed tomography. Chem Phram Bull 2002;50:609-14.
- Yamada M, Yasuhara H. Pharmacology of MAO inhibitors: safety and future. Neurotoxicology 2004;25:215-21.
- Riederer P, Danielczyk W, Grunblatt E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxiology 2004;25:271-7.
- Markianos M, Panas M, Kalfakis N, Vassilopoulos D. Platelet monoamine oxidase activity in subjects tested for Huntington抯 disease gene mutation. J Neural Transm 2004;111:475-83.
- Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods 1997;202:133-41.
- Medvedev AE, Glover V. Tribulin and endogenous MAO-inhibitory regulation in vivo. Neurotoxiology 2004;25:185-92.
- Matsukawa M, Hirai T, Karita S, Akizawa T, Hou HP, Yoshioka M, et al. A screening system of prodrugs selective for MAO-A or MAO-B. Neurotoxicology 2004;25:293-302.