
PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression1
Introduction
Hepatic fibrogenesis occurs as a wound-healing process after various chronic liver injuries, including virus infection, autoimmune liver diseases, and sustained alcohol abuse. Hepatic fibrosis eventually results in end-stage liver cirrhosis if it is not treated effectively. Hepatic stellate cells (HSC), previously known as fat- or vitamin A-storing cells or Ito cells, are the most relevant cell type for the development of hepatic fibrosis[1–3]. During liver injury, regardless of etiology, HSC become active and transdifferentiate into myofibroblast-like cells characterized by an increase in cell proliferation, loss of vitamin A-storing capability, expression of α-smooth muscle actin (α-SMA), and overproduction of extracellular matrix (ECM). Much research carried out from a therapeutic perspective has focused on searching for novel agents with inhibitory effects on HSC proliferation and activation to prevent hepatic fibrogenesis[4,5].
Cytokines and growth factors such as transforming growth factor beta (TGF-β) participate in these complicated processes, as evidenced by TGF-β-induced overproduction of extracellular matrix proteins in activated HSC[6].TGF-β acts as a potent stimulatory signal for connective tissue formation during wound repair and in fibrotic conditions, whereas the growth stimulatory effect of TGF-β appears to be mediated via an indirect mechanism involving connective tissue growth factor (CTGF, also known as hypertrophic chondrocyte-specific gene product 24 or Hcs24). CTGF, belonging to the CCN (CTGF, Cystein rich protein-Cyr61, and Nephro-blastoma overexpressed gene-Nov; CCN) family of immediate early proteins, is a 38 kD secretion polypeptide with abundant of cysteines in the C-terminal, which are involved in cell proliferation, migration and matrix production[7,8]. CTGF is specifically induced by TGF-β1 and is considered to be the downstream response element and effective molecule of TGF-β1. It has been reported that CTGF is the key regulator of extracellular matrix production and plays an important role in hepatic and renal fibrosis, thus the TGF-β1- CTGF signal pathway might be an effective target for reversal of hepatic fibrosis[9,10].
Peroxisome proliferator-activated receptors (PPAR), including α, β/δ and γ are a family of ligand-activated nuclear transcriptional factors that are emerging as important determinants of cell growth and differentiation[11]. PPARγ is present in various cells, including endothelial cells, vascular smooth muscle cells, monocytes/macrophages, and HSC. PPARγ forms a heterodimer with another nuclear receptor, retinoid X receptor α (RXRα), and the complex subsequently binds to a specific DNA sequence designated the peroxisome proliferating response element (PPRE), which is located in the promoter region of PPARγ target genes and modulates their transcription[12]. It has been demonstrated that depletion of PPARγ accompanies myofibroblastic transdifferentiation of HSC, and the treatment of activated HSC in vitro or in vivo with a variety of endogenous and exogenous PPARg ligands suppresses the fibrogenic activity of HSC. However, it is uncertain whether PPARγ is indeed a molecular target of this effect, because the ligands are also known to have receptor-independent actions, and the precise mechanisms of this inhibition have not been determined[13,14].
Based on the aforementioned information, we postulated that PPARγ might function as a countervailing factor to attenuate neocollagen formation during hepatic fibrosis, and that CTGF downregulation by PPARγ activation might be one of the mechanisms by which this occurs. To test this hypothesis, we examined the effects of activation of PPARγ on HSC growth and TGF-β1-induced CTGF expression in the present study.
Materials and methods
Reagents Recombinant TGF-β1 was purchased from Sigma (St Louis, MO, USA). 15-deoxy- Δ12,14-prostaglandin J2 (15-d-PGJ2) was the product of Cayman Chemicals (Ann Arbor, Michigan, USA). GW7845 and GW9662 were obtained from GlaxoSmithKline Pharmaceuticals (Brentford, UK). Goat anti-CTGF antibody, β-actin polyclonal antibody, and horseradish peroxidase-conjugated rabbit anti-goat IgG secondary antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Culture of HSC Rat HSC cells, kindly given to us by Prof Shi-gang XIONG (Hepatopathy Research Center of California University, USA), were resuscitated in the routine manner, resuspended with Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Hyclone, USA), 100 kU/mL penicillin G and 100 g/L streptomycin, and then planted in a 25-cm2 culture bottle and incubated in a 5% CO2 humidified atmosphere at 37 °C. The medium was changed every 3 d and the cells were trypsinized using trypsin/edetic acid when they reached 80%–90% confluence. HSC aged at passages 4–8 were used for experiments.
Lactate dehydrogenase release assay The cytotoxicity of PPARγ ligands for HSC was evaluated by using the lactate dehydrogenase (LDH) release assay. The growing HSC were treated with increasing amounts of 15-d-PGJ2 (1, 5, 10, 15, and 20 µmol/L) or GW7845 (0.1, 0.5, 1.0, 1.5, and 2.0 µmol/L) for 48 h . The LDH concentration in conditioned media was measured as medium LDH, and the LDH concentration in cell lysates was measured as cellular LDH. The concentration of LDH in DMEM with 10% FBS was defined as contamination arising from FBS and subtracted from the medium and cellular LDH concentrations. LDH activities were determined by using an LDH assay kit (Sigma). Results are presented as the percentage of total LDH=medium LDH/(medium LDH+ cellular LDH)×100%.
Cell proliferation assay The status of HSC proliferation was determined by MTT assay (Amresco, USA). Exponentially growing HSC were adjusted to 2.5×104 cells/mL with DMEM, plated in 96-well plates (Corning, USA) at 200 µL/well and then incubated for 12 h according to routine procedure. After being treated with 15-d-PGJ2 or GW7845 at various concentrations and incubated for 48 h (5 duplicate wells for each sample), 20 µL/well MTT (5 g/L) was added to each well. The medium was then removed after 4 h incubation and 100 µL/well dimethylsulfoxide (Me2SO) was added to dissolve the reduced formazan product. Finally, the plate was read in an enzyme-linked immunosorbent microplate reader (Bio-Rad 2550, USA) at 490 nm. The cellular proliferation inhibition rate (CPIR) was calculated using the following formula: CPIR=(1–average A value of experimental group/average A value of control group)×100%.
Apoptosis assay The effects of PPARγ ligands on HSC cell cycle and apoptosis were examined by flow cytometry. In brief, pretreated HSC were harvested and washed twice with phosphate-buffered saline (PBS) buffer, fixed with 70% ethanol at -20 °C for 30 min and stored at 4 °C overnight, then washed with PBS again, treated with 100 mL 100 mg/L RNase at 37 °C for 30 min and stained with 100 mL 50 mg/L Propidium Iodicle (PI) at 4 °C for 30 min in the dark. The multiplication cycle and apoptotic rate were assayed using an EPICS XL Flow Cytometer (Coulter, USA) at 488 nm, and the data were analyzed using CellQuest Software. The percentages of cells in the G0/G1 phase and S phase, and the apoptotic rate were measured by calculating the ratio of the number of corresponding cells to the number of total cells. For each sample, 10 000 cells were measured. Morphological changes resulting from apoptosis were determined by electron microscopy.
Measurement of TGF-β1-induced CTGF mRNA expression RT-PCR was performed according to the protocols recommended by TaKaRa Bio (Osaka, Japan) with some modifications. RT was performed in a total volume of 25 µL containing 3 µg total RNA (isolated by TRIzol reagent; Invitrogen, Carlsbad, CA, USA), oligo dT-adaptor primer and Avian Myeloblastosis Virus (AMV) reverse transcriptase. Primers used for amplification of CTGF cDNA were synthesized by Sangon Gene Company (Shanghai, China) with reference to the sequence described by Murphy et al[15]. The sense primer was 5'-CTA AGA CCT GTG GGA TGG GC-3' and the antisense primer was 5'-CTC AAA GAT GTC ATT GTC CCC-3'. PCR amplification yielded a PCR product of 383 bp. Primers used for amplifying a 452 bp of glyceraldehyde phosphate dehydrogenase (GAPDH) cDNA as an internal control were as follows: 5'-ACC ACA GTC CAT GCC ATC AC-3' (sense) and 5'-TCC ACC ACC CTG TTG CTG TA-3' (anti-sense). The PCR conditions were as follows: 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °Cfor 60 s. Ten microliters of the PCR product was separated on 1.5% agarose gel, stained with ethidium bromide (0.5 g/L) and quantitated by densitometry using the Image Master VDS system and associated software (Pharmacia, USA).
Western blotting analysis Cell culture medium was removed and concentrated with a Biomax Column (Millipore, Bedford, MA, USA) after TGF-β1 stimulation for 16 h. The adherent HSC were rinsed twice with cold PBS buffer, and were then harvested and lysed in ice-cold lysis buffer containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.6), 0.1% sodium dodecylsulfate (SDS), 1% Nonidet P-40, and a protease inhibitor cocktail (Boehringer Mannheim, Lewes, UK). The samples were cleared by centrifugation at 13 000×g for 10 min. Fifty micrograms of protein from cell lysate or concentrated cell medium was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene fluoride (PVDF) membranes (Immobilon, Bedford, MA, USA). After blocking in 20 mmol/L Tris-HCl (pH 7.6; containing 150 mmol/L NaCl, 0.1% Tween-20, and 5% non-fat dry milk), membranes were incubated with primary antibodies against CTGF or β-actin (used as a sample loading control) overnight at 4 °C and then incubated with horseradish peroxidase-conjugated secondary antibody. The blot was developed using the ECL detection kit (Amersham Pharmacia Biotech) according to the manufacturer’s instructions.
Morphological observations HSC were pre-fixed with glutaraldehyde at the volume fraction of 2.5% and post-fixed with 0.1% osmic acid. Subsequently, the specimens were immersed in propylene oxide after dehydration in an ethanol gradient, embedded in epoxy resin and made into ultrathin sections. Finally, the sections were stained with lead-uranium and the changes in ultrastructural organization were observed under a Hitachi-600 (Tokyo, Japan) transmission electron microscope (Figure 1).
Statistical analysis All data are expressed as mean±SD. Comparisons between groups were carried out using one-way ANOVA and the Student-Newman-Keuls q test using SPSS 11.0 (SPSS, Chicago, IL, USA). P values less than 0.05 were considered to be statistically significant.
Results
PPARγ ligands inhibited HSC proliferation Both 15-d-PGJ2 and GW7845 significantly inhibited the proliferation of HSC in a dose-dependent manner (Figure 2), for which the half effective inhibitory concentrations (IC50) were 7.38 µmol/L and 0.671 µmol/L, respectively. Correlation analysis indicated a marked positive correlation between the concentration of 15-d-PGJ2 or GW7845 and CPIR (r=0.898 and r=0.904, respectively, P<0.01). The cytotoxicity of PPARγ ligands for HSC was carefully studied by examining LDH release. As shown in Table 1, 15-d-PGJ2 and GW7845, compared with the controls, produced no significant difference in LDH release even at their highest concentrations, respectively. Moreover, after withdrawal of the PPARγ ligands, cell proliferation recovered rapidly. These results indicate that 15-d-PGJ2 and GW7845 are not toxic to cultured HSC. Therefore, 15-d-PGJ2 at 20 µmol/L and GW7845 at 2.0 µmol/L were used for the subsequent experiments.
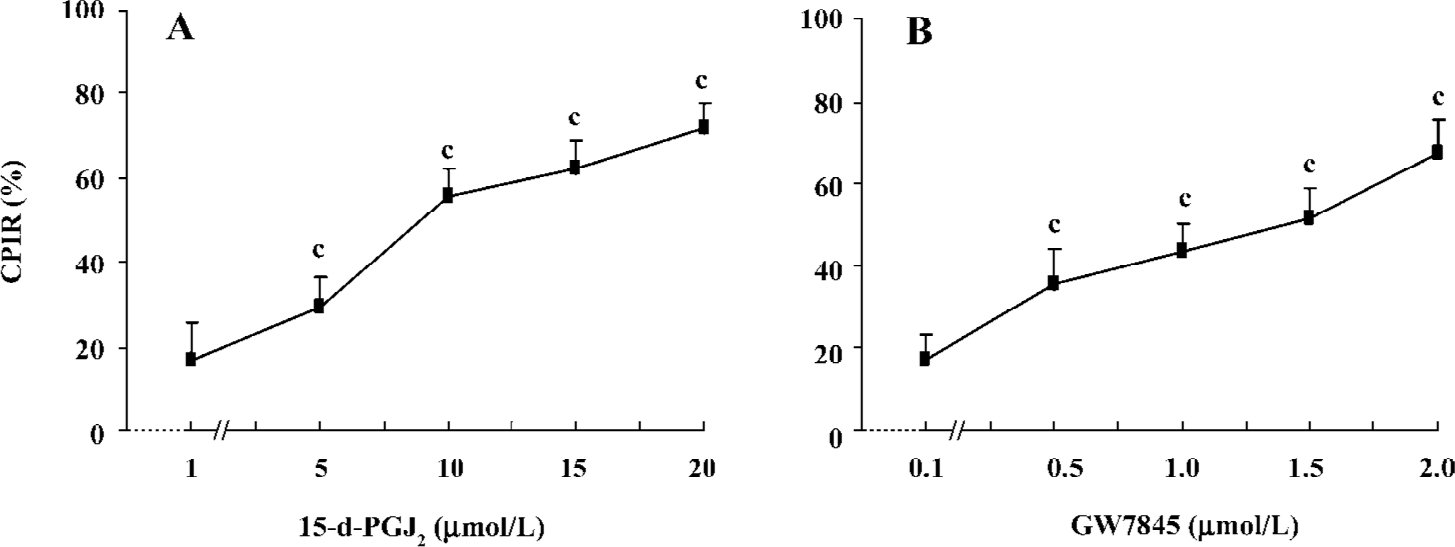
Full table
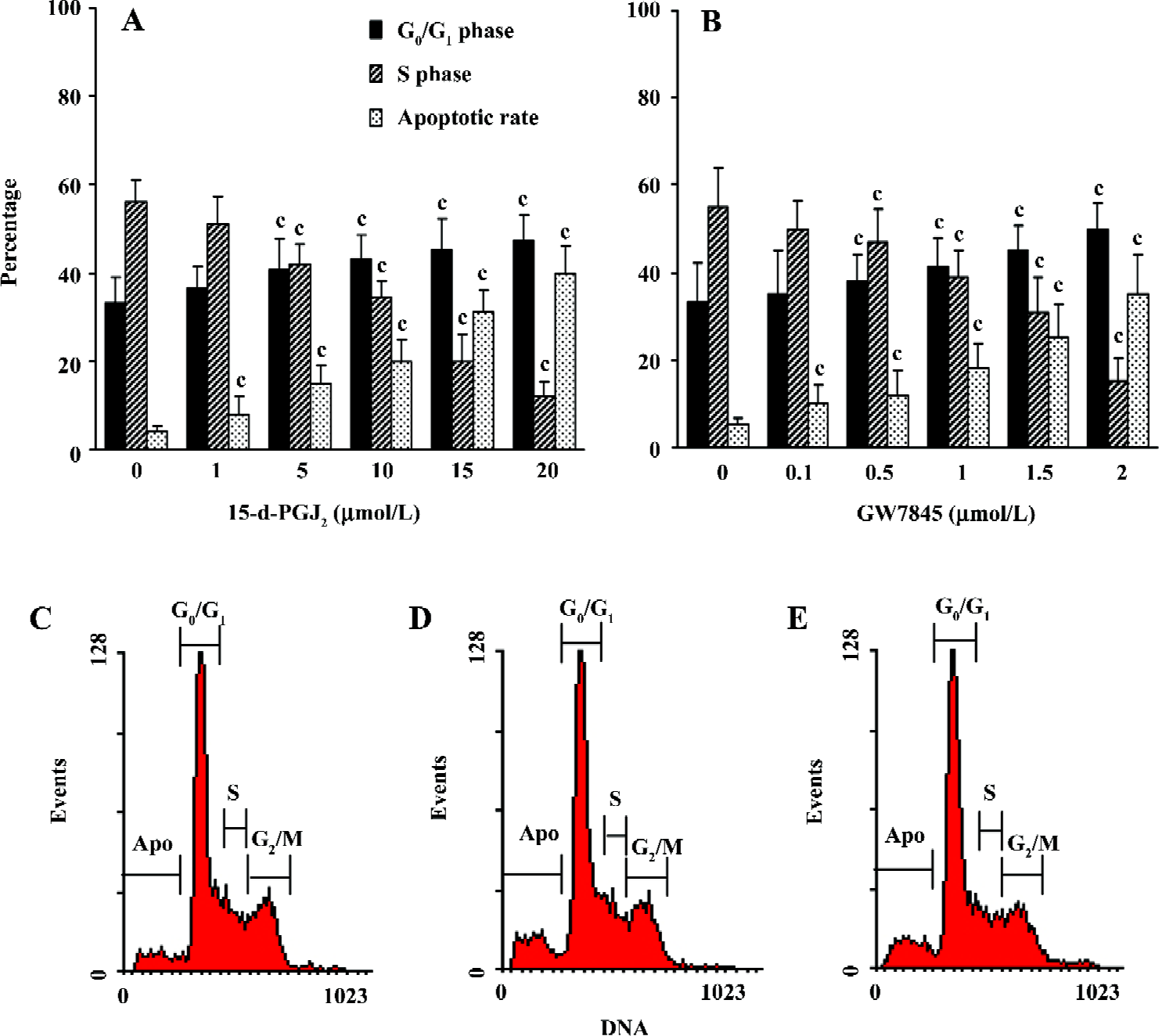
PPARγ ligands caused arrest of cell cycle and induced apoptosis in cultured HSC Cell cycle analysis of HSC was carried out by flow cytometry after exposure to various concentrations of 15-d-PGJ2 and GW7845 for 48 h. As shown in Figure 3A,3B, both 15-d-PGJ2 and GW7845 produced a dose-dependent increase in the proportion of cells in the G0/G1 phase and a decrease in the proportion of cells in the S phase. In addition, flow cytometry further demonstrated that PPARγ ligands significantly induced HSC apoptosis compared with the controls (Figure 3C–3E).
Activation of PPARγ inhibited TGF-β1-induced CTGF expression in HSC To understand the biological relevance of CTGF regulation by PPARγ, we investigated whether PPARγ activators regulate TGF-β1-induced CTGF expression in HSC. The cultured HSC were pretreated with increasing amounts of 15-d-PGJ2 or GW7845 for 1 h and were subsequently stimulated with TGF-β1 (4 ng/mL) for 4 h. The 4 ng/mL concentration and 4 h incubation time with TGF-β1 were used because our preliminary experiments indicated that these conditions yielded optimal activity (data not shown). The concentration and purity of total RNA were determined by using a DU-800 Spectrophotometer (Beckman Coulter, USA), which determined that the value of A260/A280 was between 1.6 and 1.8. Semi-quantitative RT-PCR indicated that both PPARγ activators inhibited TGF-β1-induced CTGF mRNA expression in HSC in a dose-dependent manner (Figure 4).
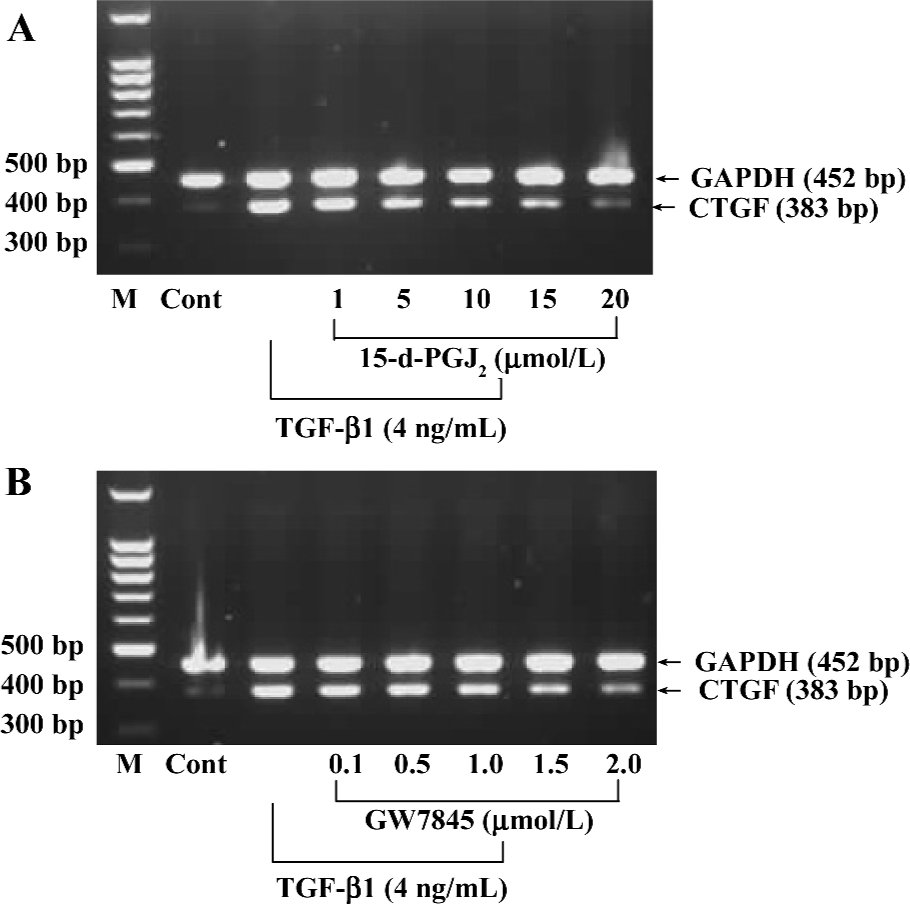
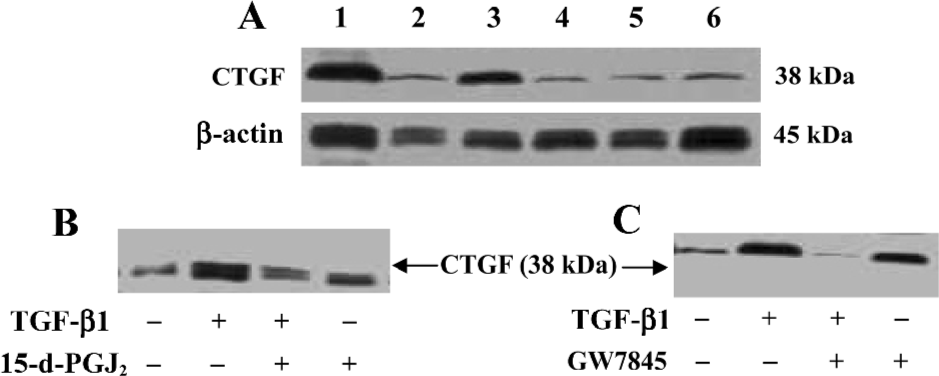
The effects of 15-d-PGJ2 and GW7845 on CTGF protein levels in TGF-β1-stimulated HSC were next examined. Consistent with previous work, we found that CTGF protein was nearly undetectable in untreated HSC by Western blotting analysis, whereas TGF-β1 significantly induced CTGF expression. Interestingly, both 15-d-PGJ2 and GW7845 dramatically inhibited TGF-β1-induced CTGF production and secretion (Figure 5). Taken together, the results indicate that PPARγ activation inhibits TGF-β1-induced CTGF expression at both the mRNA and protein levels. Moreover, the inhibitory effect is more profound and notable at the protein level than at the mRNA level, suggesting that PPARγ ligands might be exerting some translational or post-translational effects on CTGF expression. In addition, inhibition of CTGF expression was not due to cell death, because our study had proved that neither 15-d-PGJ2 nor GW7845 was toxic to HSC.
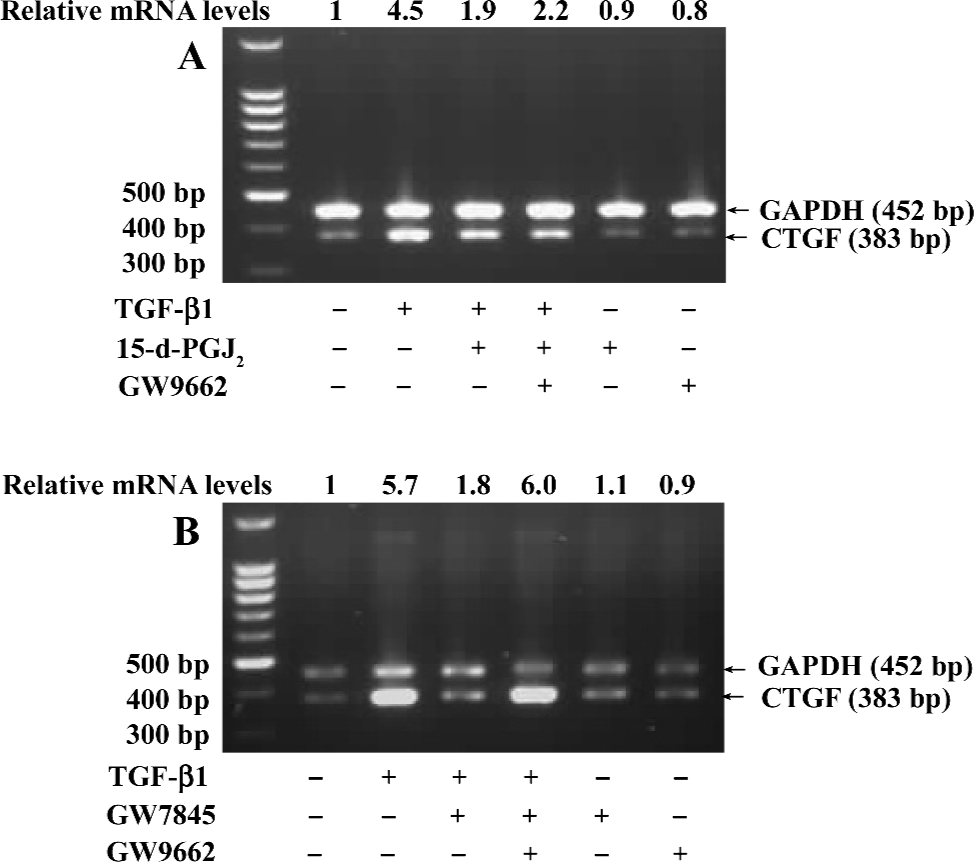
Suppression of CTGF expression was mediated by PPARγ If the suppression of TGF-β1-induced CTGF expression was indeed mediated by PPARγ, we would expect that the PPARγ-specific and irreversible antagonist GW9662 would abolish this effect. To test this hypothesis, HSC were pretreated with or without GW9662 (1 µmol/L) for 30 min prior to the addition of 15-d-PGJ2 (20 µmol/L) or GW7845 (2.0 µmol/L) and were subsequently stimulated with TGF-β1 (4 ng/mL) for 4 h. Semi-quantitative RT-PCR analysis showed that the suppression of TGF-β1-induced CTGF expression by GW7845 was almost completely abrogated by GW9662 (relative mRNA level changed from 1.8 to 6.0), whereas the inhibitory effect of 15-d-PGJ2 on CTGF mRNA expression was only partially reversed by GW9662 (relative mRNA level changed from 1.9 to 2.2; Figure 6). These data indicate that the inhibitory effect of GW7845 on TGF-β1-induced CTGF expression is largely mediated by PPARγ; however, the inhibitory effect of 15-d-PGJ2 on TGF-β1-induced CTGF expression was only mediated in part through PPARγ, suggesting that 15-d-PGJ2 could also activate some PPARγ-independent signaling pathway to repress CTGF expression in addition to the activation of PPARγ.
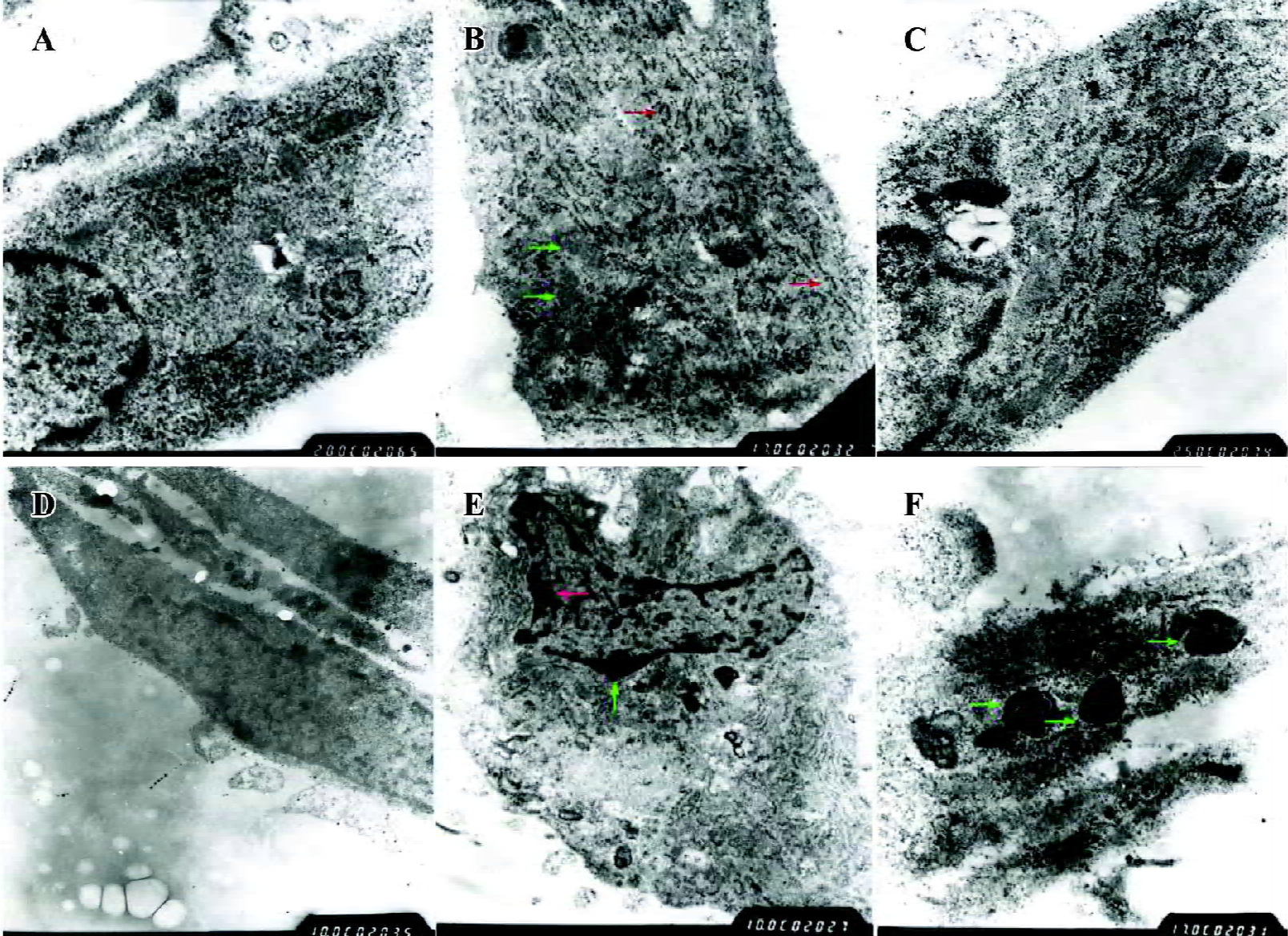
PPARγ induced a phenotypic switch from activated to quiescent HSC Unactivated HSC of the control group had typically shuttle-like or stellate forms and there were no evidently abnormal changes in the nucleus or organelles. The TGF-β1-activated HSC had enlarged cell volumes, increased amounts and dilation of the rough endoplasmic reticulum, obviously swollen mitochondria, and no intracellular lipid droplets, which were consistent with the morphological changes of HSC transdifferentiation. However, after pretreatment with PPARγ ligands, transmission electron microscopy showed that the rough endoplasmic reticulum and mitochondria were arranged in an orderly fashion, the cell membrane was complete, and intracellular lipid droplets were basically normal, which indicate that the HSC had changed from the activated to the quiescent phenotype due to PPARγ activation. Moreover, there was obvious chromatin margination and apoptotic bodies in some HSC, indicating that PPARγ ligands could also induce cell apoptosis in activated HSC (Figure 7).
Discussion
The results of the present study demonstrate that PPARγ activation inhibits HSC growth and TGF-β1-induced CTGF expression. Because activation and proliferation of HSC are critical events in the occurrence and development of hepatic fibrosis, and CTGF is a key factor in the regulation of extracellular matrix production, these repressive effects by PPARγ activation may be one of the mechanisms by which PPARγ agonists inhibit neocollagen formation during hepatic fibrosis.
It has been reported that pathological changes in hepatic fibrosis were induced in part by transcription factors that govern HSC proliferation, death, differentiation, and matrix production[16]. PPARγ is a nuclear receptor transcriptional factor and plays an important role in many biological processes, including adipogenesis, cell growth regulation, and cell differentiation. Recent studies have found that thiazolidinediones (TZD) such as pioglitazone and troglitazone, a class of anti-diabetic drugs that function as synthetic ligands of PPARγ with high affinity, inhibit neointima formation in vascular smooth muscle cells in association with decreased DNA synthesis, suggesting that PPARγ may be a potential therapeutic target for the treatment of fibrotic diseases[17–19]. In the present study, we extended these observations by determining that PPARγ activation results in the inhibition of HSC growth in a dose-dependent manner. Cell cycle status and cell apoptosis are usually closely associated; cells failing to progress to the mitosis phase are destined for apoptosis. In the past decade, emerging evidence has suggested that PPARγ ligands can induce cell apoptosis in many human malignant tumors, including breast cancer, pituitary adenomas, colon cancer, pancreatic carcinoma, and esophageal cancer[20–22]. In addition to cell cycle arrest, the inhibition of cell growth observed in HSC with PPARγ ligands may also be a result of the increase in apoptosis. In the present study, treatment with PPARγ agonists for 48 h caused G0/G1 phase arrest and blocked cells from entering the S phase. Interestingly, as seen in other tumor cells, we clearly demonstrated that PPARγ ligands induce significant apoptosis in HSC as evidenced by flow cytometry and transmission electron microscopy data. However, whether there are differences between normal cells (ie HSC) and tumor cells with respect to PPARγ quantity and intracellular distribution still remains unclear.
CTGF is a cysteine-rich mitogenic peptide that binds heparin and is secreted by fibroblasts after activation with TGF-β. CTGF is considered to function as a downstream mediator of TGF-β action on fibroblastic cells and shares some of the biological actions of TGF-β, such as the stimulation of cell proliferation and extracellular matrix protein synthesis by fibroblasts[23]. This concept is strongly supported by the fact that TGF-β over-production has been documented in nearly every fibrotic disorder, and CTGF is co-expressed at every site of fibrotic tissue formation[24]. However, CTGF does not share the growth inhibitory effect of TGF-β on epithelial cells, or appear to modulate immune or inflammatory cells[25]. These important distinctions from TGF-β suggest that CTGF could be a more desirable pharmacological target for the blockade of neocollagen formation in fibrotic disorders where TGF-β acts as an initiator. In the present study, we demonstrated that CTGF was only slightly expressed in quiescent HSC, but was dramatically upregulated by TGF-β1-stimulation, which was in accordance with the results of previous studies[26]. PPARγ activation reduced TGF-β1-induced CTGF expression at both transcriptional and post-transcriptional levels, and activated HSC had significant morphological differences in the activated and quiescent phenotypes, thus the PPARγ-CTGF pathway might be a target for a novel antifibrotic strategy.
Recently, it has been reported that there exist PPARγ-independent effects of PPARγ ligands at high doses[27]. In addition, the PPARγ natural ligand 15-d-PGJ2 has many functions other than that of a PPARγ activator[28]. In the present study, we showed that the high affinity PPARγ ligand GW7845 could inhibit HSC growth and TGF-β1-induced CTGF expression. By using the PPARγ-specific antagonist GW9662, we demonstrated that the inhibitory effect of GW7845 on TGF-β1-induced CTGF expression was mediated by PPARγ. However, the inhibitory effect of 15-d-PGJ2 on TGF-β1-induced CTGF expression was partly due to PPARγ activation, suggesting that 15-d-PGJ2 can activate other PPARγ-independent signaling pathways to repress CTGF expression. Sequence analysis of the CTGF promoter revealed that there are two putative NF-κB sites, two putative AP-1 sites, and a putative Smads binding element (SBE). The activated-PPARγ–RXRα complex could bind to SBE, also known as a PPRE, to inhibit CTGF gene transcription[29,30]. Interestingly, it has been reported that 15-d-PGJ2 inhibits target gene expression not only by interference with Smads, but also by directly inhibiting NF-κB, from which it could be inferred that the inhibition of 15-d-PGJ2 on TGF-β1-induced CTGF expression was only partly abrogated by GW9662[31].
In conclusion, our studies demonstrated that PPARγ activation can inhibit HSC proliferation and induce cell apoptosis. Furthermore, PPARγ ligands markedly inhibited TGF-β1-induced CTGF expression in HSC. Because the biological actions of TGF-β are complicated and affect many different cell types, whereas CTGF is mainly produced and secreted by activated HSC, CTGF may serve as a more specific target for selective intervention in processes involving connective tissue formation during hepatic fibrosis. A better understanding of PPARγ could lead to the development of a new therapeutic approach for the control and reversion of hepatic fibrosis in humans. However, it should be emphasized that the results in the present study were generated from cultured HSC and that they might not necessarily and comprehensively reflect the behavior of quiescent HSC in vivo. Further experiments, beyond the scope of the present study, are required to elucidate the underlying mechanisms of PPARγ in the inhibition of HSC proliferation.
References
- She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 2005;280:4959-67.
- Lee SH, Seo GS, Park YN, Sohn DH. Nephroblastoma over-expressed gene (NOV) expression in rat hepatic stellate cells. Biochem Pharmacol 2004;68:1391-400.
- Mann DA, Smart DE. Transcriptional regulation of hepatic stellate cell activation. Gut 2002;50:891-6.
- Friedman SL. Liver fibrosis: from bench to bedside. J Hepatol 2003;38:38-53.
- Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol 2002;36:200-9.
- Shin JY, Hur W, Wang JS, Jang JW, Kim CW, Bae SH, et al. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-beta1. Exp Mol Med 2005;37:138-45.
- Kobayashi H, Hayashi N, Hayashi K, Yamataka A, Lane GJ, Miyano T. Connective tissue growth factor and progressive fibrosis in biliary atresia. Pediatr Surg Int 2005;21:12-6.
- Gao R, Ball DK, Perbal B, Brigstock DR. Connective tissue growth factor induces c-fos gene activation and cell proliferation through p44/42 MAP kinase in primary rat hepatic stellate cells. J Hepatol 2004;40:431-8.
- Uchio K, Graham M, Dean NM, Rosenbaum J, Desmouliere A. Down-regulation of connective tissue growth factor and type I collagen mRNA expression by connective tissue growth factor antisense oligonucleotide during experimental liver fibrosis. Wound Repair Regen 2004;12:60-6.
- Perbal B. CCN proteins: multifunctional signalling regulators. Lancet 2004;363:62-4.
- Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol 2003;285:20-30.
- Sung CK, She H, Xiong S, Tsukamoto H. Tumor necrosis factor-alpha inhibits peroxisome proliferator-activated receptor gamma activity at a posttranslational level in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2004;286:722-9.
- Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K, et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem 2004;279:11392-401.
- Planaguma A, Claria J, Miquel R, Lopez-Parra M, Titos E, Masferrer JL, et al. The selective cyclooxygenase-2 inhibitor SC-236 reduces liver fibrosis by mechanisms involving non-parenchymal cell apoptosis and PPAR gamma activation. FASEB J 2005;19:1120-2.
- Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, Martin F, et al. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem 1999;274:5830-4.
- Kinnman N, Francoz C, Barbu V, Wendum D, Rey C, Hultcrantz R, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest 2003;83:163-73.
- Giannini S, Serio M, Galli A. Pleiotropic effects of thiazolidine-diones: taking a look beyond antidiabetic activity. J Endocrinol Invest 2004;27:982-91.
- Enomoto N, Takei Y, Hirose M, Konno A, Shibuya T, Matsuyama S, et al. Prevention of ethanol-induced liver injury in rats by an agonist of peroxisome proliferator-activated receptor-gamma, pioglitazone. J Pharmacol Exp Ther 2003;306:846-54.
- Wang MY, Unger RH. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am J Physiol Endocrinol Metab 2005;288:216-21.
- Yang FG, Zhang ZW, Xin DQ, Shi CJ, Wu JP, Guo YL, et al. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol Sin 2005;26:753-61.
- Heaney AP, Fernando M, Melmed S. PPAR-gamma receptor ligands: novel therapy for pituitary adenomas. J Clin Invest 2003;111:1381-8.
- Fauconnet S, Lascombe I, Chabannes E, Adessi GL, Desvergne B, Wahli W, et al. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J Biol Chem 2002;277:23534-43.
- Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol Res 2003;26:1-9.
- Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem 2004;279:8848-55.
- Kurikawa N, Suga M, Kuroda S, Yamada K, Ishikawa H. An angiotensin II type 1 receptor antagonist, olmesartan medoxomil, improves experimental liver fibrosis by suppression of proliferation and collagen synthesis in activated hepatic stellate cells. Br J Pharmacol 2003;139:1085-94.
- Gao R, Brigstock DR. Low density lipoprotein receptor-related protein (LRP) is a heparin-dependent adhesion receptor for connective tissue growth factor (CTGF) in rat activated hepatic stellate cells. Hepatol Res 2003;27:214-20.
- Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, et al. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology 2000;31:101-8.
- Wakino S, Hayashi K, Kanda T, Tatematsu S, Homma K, Yoshioka K, et al. Peroxisome proliferator-activated receptor gamma ligands inhibit Rho/Rho kinase pathway by inducing protein tyrosine phosphatase SHP-2. Circ Res 2004;95:45-55.
- Ozaki S, Sato Y, Yasoshima M, Harada K, Nakanuma Y. Diffuse expression of heparan sulfate proteoglycan and connective tissue growth factor in fibrous septa with many mast cells relate to unresolving hepatic fibrosis of congenital hepatic fibrosis. Liver Int 2005;25:817-28.
- Hsu YC, Lin YL, Chiu YT, Shiao MS, Lee CY, Huang YT. Antifibrotic effects of Salvia miltiorrhiza on dimethylnitrosamine-intoxicated rats. J Biomed Sci 2005;12:185-95.
- Han YP, Zhou L, Wang J, Xiong S, Garner WL, French SW, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem 2004;279:4820-8.