
Alterations of placental cytochrome P450 1A1 and P-glycoprotein in tobacco-induced intrauterine growth retardation in rats1
Introduction
There is ample epidemiological evidence that individuals with intrauterine growth retardation (IUGR) carry an increased risk of prenatal morbidity and mortality[1] and that those born with IUGR are at a much greater risk of a wide range of medical problems, such as mental handicaps and neurobehavioral disorders[2,3]. There are various possible causes of pathologically induced IUGR, and it is reported that some xenobiotic exposure during pregnancy, including to tobacco and drugs, could be attributable to IUGR[4,5]. Clinically, there is no generally accepted effective treatment at present and, therefore, the understanding of the mechanisms for controlling fetal growth and the causes for IUGR induced by xenobiotics is extremely important in the development of therapeutic options.
The placenta is the organ responsible for the transfer of nutrients and waste products between the fetal and maternal circulations, and plays a pivotal role in fetal growth[6]. The placenta receives oxygen from the maternal circulation and is positioned in an oxygen gradient between the mother and fetus. The placenta provides a link between the circulations of 2 distinct individuals, but also acts as a barrier to protect the fetus from xenobiotics in the maternal blood. Animal studies, such as embryo transplant and cross breeding experiments, have shown that, unlike postnatal growth, the growth of the fetus is controlled predominately by the uterine environment, and not by fetal or maternal genetic factors[7]. Therefore, placental dysfunction can have an important effect on fetal intrauterine development and can lead to fetal diseases such as IUGR.
The placenta can perform xenobiotic transportation and biotransformation. The transfer of foreign chemicals across the placenta can be modified by metabolism in the placenta itself. The extent to which drugs cross the placenta is also modulated by the actions of placental phase I and II drug-metabolizing enzymes. Cytochrome P450 enzymes in particular have been well characterized in the placenta at the level of mRNA, protein, and enzyme activity[8]. In some cases, however, the enzymes can activate exogenous compounds, making them toxic to the fetus[6]. Cytochrome P450 1A1 (CYP1A1) exists mainly in extra-hepatic tissue, and can activate some xenobiotics with possible deleterious effects. The alteration in CYP1A1 expression, for example, as a result of inducers/inhibitors or maternal diseases, could potentially adversely affect placental function and pregnancy outcome[9]. Studies show that CYP1A1 expresses in the placentas of women who are exposed to cigarettes[6]. However, little is known about the possible changes of placental CYP1A1 expression over the course of pregnancy and its roles in IUGR formation.
Some drugs are pumped across the placenta by various active transporters located on both the fetal and maternal side of the trophoblast layer, and instances of drug-induced birth defects have been in part blamed on the placenta’s apparent ‘leakiness’ to maternal blood-borne agents[8]. The impact of active transporters such as P-glycoprotein (P-gp) on the disposition of drugs has been demonstrated in some studies[10–12]. P-gp, which is encoded by the mdr gene, is a membrane transport protein that functions as an efflux pump for various cytotoxic compounds and, therefore, reduces the intracellular concentrations of these compounds[10]. Studies suggest that P-gp of the trophoblast cells is involved in the function of the blood–placental barrier and is necessary in reducing fetal drug exposure[11,12], although little is known about the expression of placental P-gp in different gestational time and its significance to IUGR.
It is difficult to correlate toxic chemicals and their transportation and metabolism in the human placenta with IUGR. The rat placenta is morphologically and histologically similar to the human one; both of them are of the same hemochorial type. Therefore, a rat model of IUGR established by tobacco smoking is useful in providing us with information about the relationships among xenobiotics, placental transportations/metabolisms, and IUGR. To understand the toxic mechanism of tobacco in IUGR formation, in the present study, the alterations of placental CYP1A1 and P-gp expressions were investigated in IUGR rats, so as to explore the possible significance of placental CYP1A1 and P-gp in tobacco-induced IUGR.
Materials and methods
Chemicals Thiobarbituric acid (TBA) and 5, 5'-dithio-2, 2'-dinitrobenzoic acid were obtained from Sigma. Trizol reagent was obtained from Molecular Research Center and One Step RNA PCR Kit was obtained from TaKaRa Biotechnology. Oligonucleotide primers were custom synthesized by Sangon Biological Engineering Technology. Rabbit anti-rat mdr1 and streptavidin-peroxidase (SP) reagent were obtained from BOSTER Biotechnology. All other chemicals and reagents were of analytical grade.
Animals Specific pathogen free(SPF) Wistar rats with the weights of 190±18 g (female)/280±23 g (male) were obtained from the Experimental Center of Medical Scientific Academy of Hubei (China, No 2003-0005). Virgin female and male rats were left undisturbed for 5 d and then subjected to experimental conditions. Overnight, every 2 females was mated up with 1 male rat and the occurrence date of a vaginal plug was considered as gestational d 0 (GD 0). Pregnant rats were then housed 1 per cage in an environment of constant temperature (21±2 °C) and relative humidity (50%±10%) with a 12 h L:D cycle. Ad libitum access to a standard diet and water was permitted. The study protocol was in accordance with the guidelines for animal research and was approved by the Ethical and Research Committee of Wuhan University.
Tobacco smoke exposure On GD 7, pregnant rats were allocated to either a control group or a tobacco group. A rat IUGR model was processed, with modification, according to the procedure of Younoszai and Li[13,14]. From GD 7 till parturition (GD 21), at 8:00, 11:00, 14:00, and 17:00, four pregnant rats from the tobacco group were subjected to cigarette smoke in a chamber measuring 40 cm×28 cm×18.5 cm for 15 min, with the gross smoke concentration set at 9 g·m-3. The tar and nicotine yields of commercial cigarettes were 15 and 1.1 mg per cigarette, respectively. The control group was sham-treated; that is, a group of 4 pregnant rats were in the same chamber without cigarette smoke for 15 min. The pregnant rats were weighed at GD 0, GD 7, GD 14, and GD 21. Animals were killed at 8:00 on GD 14 and GD 21, corresponding to mid and late gestational time[15]. Each feto-placental unit was removed quickly from the uterus and placenta specimens were excised, rinsed with cold saline, immediately frozen in liquid nitrogen and stored at -80°C until used. Placenta weight and fetal development parameters (fetal body and organ weights, and fetal body and tail lengths) were recorded.
Placental pathomorphological observations and immunohistochemistry Fresh placentas were put into 4% paraformaldehyde in phosphate buffered saline (PBS) for 24 h. The fixed placental samples were processed using standard histological techniques and stained with hematoxylin and eosin. The slides were observed using light microscopy (Axiopstar PLUS).
Routine immunohistochemistry SP method was carried out. The paraffin slides were deparaffinized with xylene and rehydrated in a graded series of ethanol. To quench endogenous peroxidase activity 0.3% H2O2 was added for 10 min, and preimmune goat serum was used to block non-specific binding sites. Sections were then incubated at 37 °C for 20 min with a rabbit polyclonal anti-mdr1 antibody diluted in PBS (1:100). The slides were then incubated at 37°C for 15 min with an anti-rabbit secondary antibody and visualized with a DAB chromatogen system. PBS substituted for anti-mdr1 antibody for negative staining control. The slides were observed under light microscopy (Axiopstar PLUS) and the average gray level was measured using Photo Imaging System (HMIAS-2000). The quantitative stereology was performed in triplicates with 5 fields in each slide.
Tissue biochemical analysis Placentas at GD 21 were homogenized with saline to be 10% (w/v) homogenates. After the homogenates were centrifuged at 200×g for 10 min, the supernatants were centrifuged at 9 000×g for 20 min and, finally, collected and stored at -30 °C for the further assays. Placental protein concentrations were determined using the Lowry method[16], with the bovine serum albumin (BSA) as standard. The extent of lipid peroxidation was detected by measuring malondialdehyde (MDA) content using TBA according to a modified procedure described by Ondrejickova et al[17]. The activities of superoxide dismutase (SOD) and catalase (CAT) were measured as described above[18,19].
Preparation of placental total RNA Total RNA was isolated from the frozen placentas according Trizol reagent instructions. The protocol involved disruption of cells, denaturation of nucleoprotein complexes, inactivation of endogenous ribonuclease (RNase) activity and, finally, removal of proteins and degradation of residual DNA by nuclease digestion. The concentration and purity of RNA were determined using a spectrophotometer (UV-1601, Shimadzu) and adjusted to 1 µg/µL. Total RNA was stored in DEPC-H2O at -80 °C until used.
Semiquantitative reverse transcriptase-polymerase chain reaction The cDNA synthesis and polymerase chain reaction (PCR) amplification were produced in 1 step using Promega’s reverse transcriptase-polymerase chain reaction (RT-PCR) System. Different primers and PCR products are shown in Table 1. The final concentrations of reagents in the RT-PCR reaction system were as follows: 1×One Step RNA PCR buffer, 5 mmol/L MgCl2, 1 mmol/L of each dNTP, 0.8 U/µL RNase inhibitor, 0.1 U/µL avian myeloblastosis virus (AMV) RTase XL, 0.1 U/µL AMV-optimized Taq, 0.4 µmol/L of each primer, and 0.02 U/µL placenta RNA in each 50 µL reaction volume. For quantitative analysis of mRNA expression, the housekeeping gene cyclophilin, an internal loading control[22], was used to amplify together with the specific target gene in 1 tube. RT-PCR reactions were carried out in a thermal cycler: 50 °C for 30 min for reverse transcription, then 94 °C, 2 min for RT inactivation; finally 72 °C, 5 min for a terminal elongation step following the amplification cycles. PCR cycling conditions were as follows: 94 °C, 30 s; 62 °C, 45 s; 72 °C, 30 s, 45 cycles for CYP1A1. 94 °C, 30 s; 54 °C, 60 s; 72 °C, 30 s, 30 cycles for mdr1a. 94 °C, 30 s; 56 °C, 20 s; 72 °C, 30 s, 25 cycles for mdr1b. DEPC-H2O substituted for placental RNA for negative control. An aliquot (4 µL) of the RT-PCR reactions was separated on a 1.5% agarose gel containing ethidium bromide, visualized under UV light, photographed, and analyzed by densitometry using Photo Documentation and Imaging System (Bio-1D). The expression level of target gene mRNA was shown as the ratio of the intensities of the target-specific band and the CYC band individually.
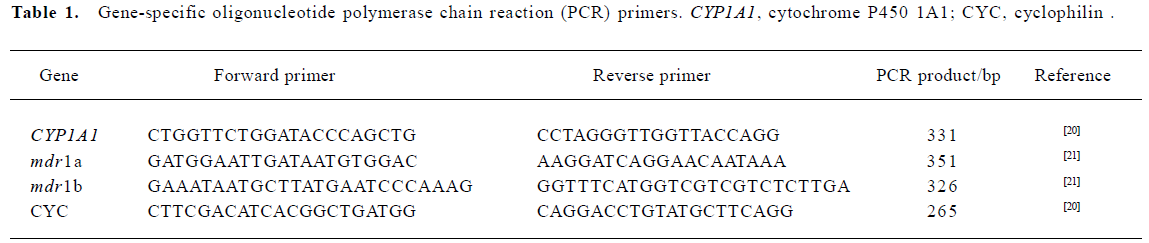
Full table
Statistical analysis The experimental results were expressed as mean±SD. Statistical Packages for Social Sciences (SPSS) was used for data analysis. Analysis of variance (ANOVA) was used for comparison of means of several groups and χ-square analysis was performed to test for differences in proportions of categorical variables between 2 groups. The level of significance was set at P< 0.05.
Results
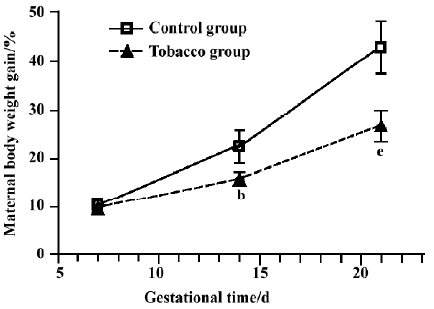
Maternal body weights The change in pregnant rat body weight is a useful, indirect indicator of physical development. During the period of GD 7 to GD 21, there was a significant decline in the body weight and percentage weight gain after tobacco exposure (Figure 1).
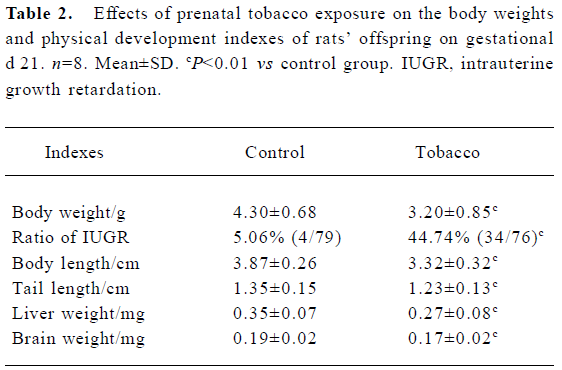
Neonatal body weights and physical development indexes Body weight was an important index for diagnosing IUGR (IUGR was diagnosed by the standard that the mean body weight in the treated group was less 2 standard deviations than that in the control group)[23]. The offspring in the tobacco group showed a lower average body weight (3.20 g) than the control group (4.30 g) at GD 21, indicating fetal IUGR. The ratio of IUGR increased to 44.74% (34/76) in the tobacco group, whereas it was only 5.06% (4/79) in the control group. In addition, the body and tail lengths, and liver and brain weights of the tobacco group significantly lagged behind those of control group at GD 21, with a suppression of14.2%, 8.9%, 22.9%, and 10.5%, respectively (P<0.01) (Table 2).
Full table
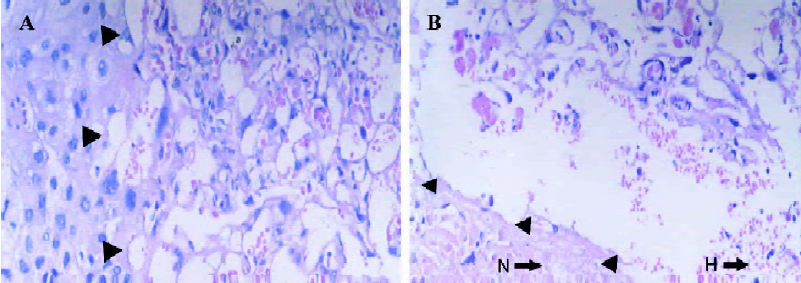
Placental pathomorphology The rat placenta is composed of 3 distinct zones: labyrinth, basal, and maternal deciduas; where the labyrinth zone represents the main area of maternal-fetal exchange. According to our results, compared to those in the control group (Figure 2A), the obvious changes occurred in the labyrinth and basal deciduas of the full-term placenta in IUGR rats, such as through interstitial and endovascular hemorrhage in the labyrinth zone, and trophoblast cells necrosis in the basal zone (Figure 2B). Full-term placental weight (0.39±0.08 g) of tobacco-induced IUGR rats decreased by more than that of the normal placentas (0.49±0.06 g).
Placental antioxidative system In the tobacco exposure group, placental MDA content was significantly increased by 50.0% (P<0.05), and the activities of SOD and CAT were notably decreased by 58.2% and 30.6% (P<0.05, P<0.01), respectively (Table 3).
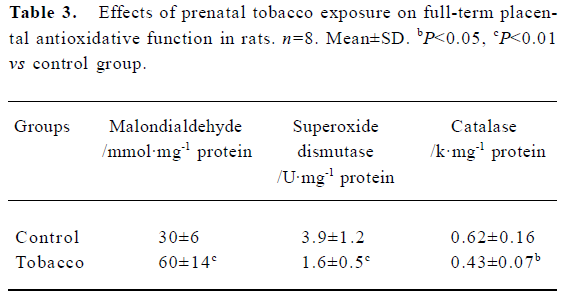
Full table
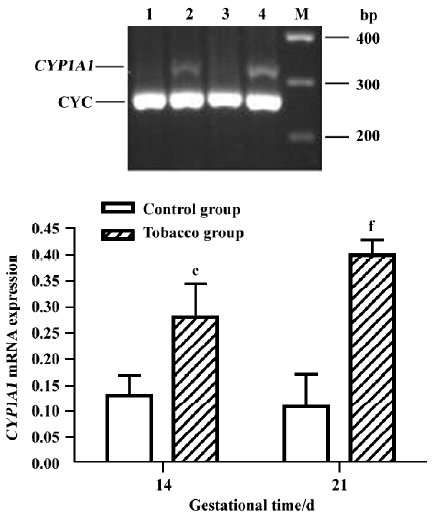
Placental CYP1A1 mRNA expression No PCR product for CYP1A1 mRNA was detected in normal placentas. However, expression was detected in the placentas of the tobacco exposure group; furthermore, the level at GD 21 was approximately 1.39-fold higher than that at GD 14, with a faint band (Figure 3).
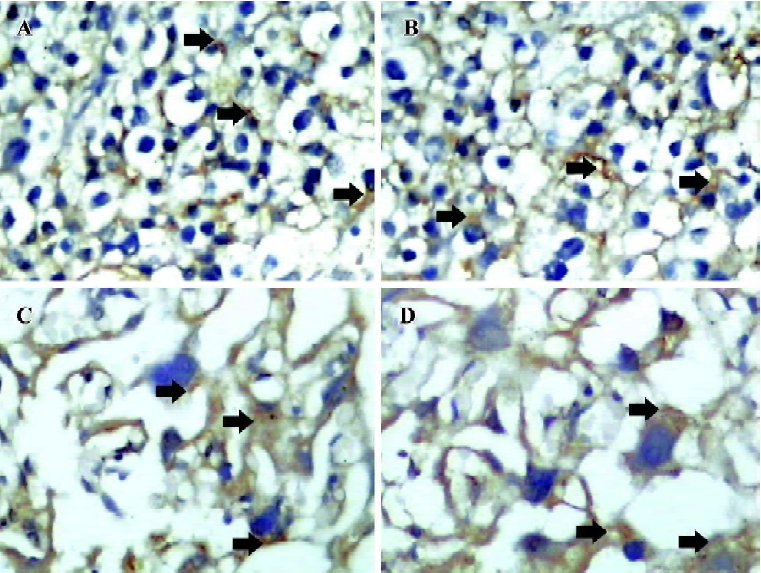
Placental P-gp and mdr1 mRNA expression Immunohistochemical analysis with anti-mdr specific antibody showed strong positive staining in placenta trophoblasts (Figure 4A–D). The P-gp level at GD 14 in the tobacco group (Figure 4B) was similar to that of the normal placenta (Figure 4A). However, the level at GD 21 in the tobacco group (Figure 4D) was higher than that of normal placenta (Figure 4C), almost 1.24-fold higher (P<0.05).
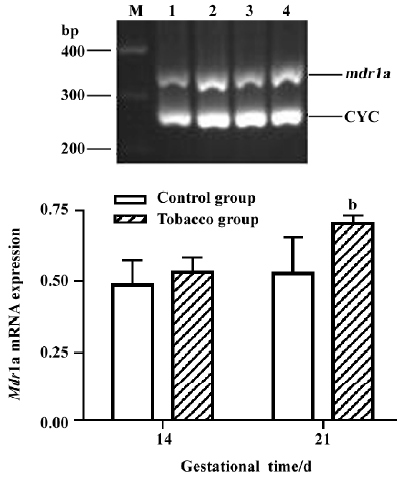
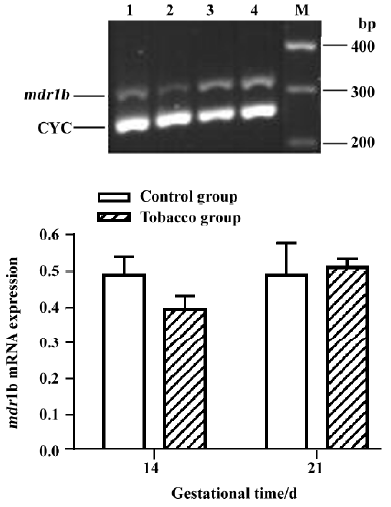
The levels of mdr1a mRNA at GD 14 and GD 21 in the placentas of tobacco groups were approximately 1.10-fold and 1.32-fold higher than those in the normal placentas (Figure 5). mdr1b mRNA level, however, was not significantly different between the 2 groups (Figure 6).
Discussion
The placenta, which is derived from both fetal and maternal tissues, is considered the first fetal organ to be exposed to exogenous substances and plays an important role in fetal intrauterine development. In the present study, gestational tobacco smoking resulted in significantly delayed fetal growth, as indicated by the fetal development parameters, especially fetal body weight. Meanwhile, placental weight decreased and obvious pathomorphological changes were observed. Our result showed that the content of MDA was higher and the activities of SOD and CAT were lower in IUGR placentas, suggesting a decreased ability of placental antioxidative defense. Therefore, these results suggest that prenatal tobacco exposure has a specific and deleterious effect on the placenta that secondarily limits fetal growth.
The placenta is now viewed as a metabolic barrier rather than a physical barrier[24]. The placenta can perform biotransformation that occurs in the liver. Numerous foreign compounds reach the placenta through the maternal circulation and placental tissue is capable of oxidizing several of them. In present study, the expression of CYP1A1 mRNA was detected in the placentas of the tobacco group, but not in the normal placentas; furthermore, the expression of CYP1A1 mRNA was higher in full-term placentas than that in mid-term placentas. These results demonstrate that an increase of CYP1A1 with environmental tobacco exposure is obvious and that a time-response relationship exists during tobacco consumption. There are many well-known CYP1A1 inducers in tobacco, and smoking is one important source of exposure to them. Increased CYP1A1 level might explain the toxic mechanism because polyarylhydrocarbons can be bioactivated by CYP1A1 and can generate reactive metabolites[25]. Wu et al found that placental and fetal tissues were capable of metabolizing benzo(a)pyrene [B(a)P], albeit to a lower extent, and that CYP1A1 was involved in metabolism of inhaled B(a)P[26]. In addition, CYP1A1 can result in free radical formation and lipid peroxidation, and then affect uterine redox environment and fetal growth. Oxidative stress has been increasingly postulated as a major contributor to dysfunction in IUGR[27], and our data indicates a decreased ability of placental antioxidative defense. The present finding of significant upregulation of placental CYP1A1 mRNA subsequent to prenatal tobacco exposure supports a hypothesis that an elevated placental CYP1A1 level can be a trigger for IUGR.
In P-gp, generally, mdr1a appeared to be the pharmacologically most relevant isotype. Schinkel et al also indicated that mdr1a was the critical placental P-gp in mice[28]. In a study by Lin, Mdr1b was highly induced in the secretary epithelium endometrium of the uterus in mice during pregnancy and might protect steroid secreting cells from potentially damage[29]. In the present study, we analyzed P-gp expression in the rat placenta during its maturation, using the methods of immunohistochemistry. Intensive immunoreactivity for P-gp was found in the developing labyrinth zone of normal placentas. Furthermore, tobacco smoking induced P-gp expression of the trophoblast cells only in full-term placentas. We examined the changes of mdr1 levels in different stages, using the RT-PCR method. We found different P-gp members displayed distinct inductive patterns. Our PCR data indicates that the expression of mdr1a gene tends to increase from GD 14 to GD 21 in the normal placenta, which correlates well with the results of Novotna et al[30]. During tobacco smoke exposure, a significant increase in the amount of mdr1a mRNA has been observed at GD 21, although it remained steady at GD 14. The level of mdr1b almost remained steady at GD 14 and GD 21 between control and tobacco groups. These results suggest that tobacco smoke induced P-gp, especially mdr1a. The different expressional patterns of mdr1a and mdr1b genes show that they were not co-regulated during pregnancy and signified that the expression of both genes probably underlied different regulation pathways, which is supported by the recent data of Lee[31,32]. P-gp was able to extrude a wide variety of structurally and chemically unrelated compounds out of cells[33]. Researches have selected B(a)P as a potential P-gp substrate[34]. There-fore, we suspect that the higher expression of P-gp in rat IUGR placenta might be caused by the pumping out of part of the tobacco toxins, to decrease toxicity and, thus, to protect the fetus from potentially harmful xenobiotics during tobacco exposure, which might be a compulsory mechanism of IUGR. However, we cannot exclude the possibility that there exist some P-gp inducers in tobacco. Further experiments are needed to examine this possibility.
In summary, the present study has discussed the abnormal alterations of placental morphology and function in tobacco-induced IUGR. The expression of placental CYP1A1 and P-gp increased in tobacco-induced IUGR. Overexpres-sion of placental CYP1A1 can contribute to the metabolism of tobacco and can generate reactive metabolites, which can be a trigger for IUGR. However, upregulation of P-gp might pump out part of the tobacco toxins, as a compulsory mechanism of IUGR, to decrease tobacco exposure to the developing fetus.
References
- Genbacev O, McMaster MT, Zdravkovic T, Fisher SJ. Disruption of oxygen-regulated responses underlies pathological changes in the placentas of women who smoke or who are passively exposed to smoke during pregnancy. Reprod Toxicol 2003;17:509-18.
- Amin H, Singhal N, Sauve RS. Impact of intrauterine growth restriction on neurodevelopmental and growth outcomes in very low birth weight infants. Acta Paediatr 1997;86:306-14.
- Spinillo A, Stronati M, Ometto A, Fazzi E, Lanzi G, Guaschino S. Infant neurodevelopmental outcome in pregnancies complicated by gestational hypertension and intra-uterine growth retardation. J Perinat Med 1993;21:195-203.
- Peacock JL, Cook DG, Carey IM, Jarvis MJ, Bryant AE, Anderson HR, et al. Maternal cotinine level during pregnancy and birth weight for gestational age. Int J Epidemiol 1998;27:647-56.
- Chiaffarino F, Parazzini F, Paladini D, Acaia B, Ossola W, Marozio L, et al. A small randomised trial of low-dose aspirin in women at high risk of pre-eclampsia. Eur J Obstet Gynecol Reprod Biol 2004;112:142-4.
- Pasanen M, Pelkonen O. The expression and environmental regulation of P450 enzymes in human placenta. Crit Rev Toxicol 1994;24:211-29.
- Holmes R, Montemagno R, Jones J, Rodeck C, Soothill P. Fetal and maternal plasma insulin-like growth factors and binding proteins in pregnancies with appropriate or retarded fetal growth. Early Hum Dev 1997;49:7-17.
- Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet 2004;43:487-514.
- Pasanen M. The expression and regulation of drug metabolism in human placenta. Adv Drug Deliv Rev 1999;38:81-97.
- Islam OM, Hara M, Miyake J. Induction of P-glycoprotein, glutathione-S-transferase and cytochrome P450 in rat liver by atrazine. Environ Toxicol Pharmacol 2002;12:1-6.
- Lankas GR, Wise LD, Cartwright ME, Pippert T, Umbenhauer DR. Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod Toxicol 1998;12:457-63.
- Smit JW, Huisman MT, Tellingen O, Wiltshire HR, Schinkel AH. Absence or pharmacological blocking of placental P-glycoprotein profoundly increases fetal drug exposure. J Clin Invest 1999;104:1441-7.
- Younoszai MK, Peloso J, Haworth JC. Fetal growth retardation in rat exposed to cigarette tobacco during pregnancy. Am J Obstet Gynecol 1969;104:1027.
- Li Y, Wang H, Li JF. In utero exposure to tobacco and alcohol modifies neurobehavioral development in mice offspring: Consideration a role of oxidative stress. Pharmacol Res 2004;49:467-73.
- Kenagy J, Liu F, Soares M, Audus KL. Gestational and tobacco effects on peptidase activity in the placenta. Peptides 1998;19:1659-66.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-75.
- Ondrejickova O, Horakova L, Juranek I, Ziegelhoeffer A, Stolc S. Effect of stobadine on lipid peroxidation in brain and heart after ischemia and reperfusion of the brain. Life Sci 1999;65:1959-61.
- Wang H, Peng RX, Kong R, Zeng HG. Effect of allicin on ethanol-induced hepatotoxicity in mice. Wei Sheng Yan Jiu 1998;27:415-7.
- Wang H, Peng RX, Wang RK, Kong R. Antagonizing effect of sodium ferulate on the changes of hepatic antioxidative function induced by ethanol in mice. Acta Pharm Sin 1997;32:511-4.
- Morris DL, Davila JC. Analysis of rat cytochrome P450 isoenzyme expression using semi- quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Biochem Pharmacol 1996;52:781-92.
- Kwan P, Sills GJ, Butler E, Gant TW, Brodie MJ. Differential expression of multidrug resistance genes in naive rat brain. Neurosci Lett 2003;339:33-6.
- Cavicchioli L, Flanigan TP, Dickson JG, Vantini G, Dal Toso R, Fusco M, et al. Choline acetyltransferase messenger RNA expression in developing and adult rat brain: Regulation by nerve growth factor. Brain Res Mol Brain Res 1991;9:319-25.
- Chatelain P. Children born with intra-uterone growth retardation (IUGR) or small for gestational age (SGA): Long term growth and metabolic consequences. Endocr Regul 2000;34:33-6.
- Gupta RC, Sastry BV. Toxicology of the placenta. In: Ballantyne B, Marrs TC, Syversen T, editors. General and applied toxicology. London: MacMillan; 2000. p 1233–63.
- Whitlock JP. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol 1999;39:103-25.
- Wu J, Ramesh A, Nayyar T, Hood DB. Assessment of metabolites and AhR and CYP1A1 mRNA expression subsequent to prenatal exposure to inhaled benzo(a)pyrene. Int J Dev Neurosci 2003;21:333-46.
- Takagi Y, Nikaido T, Toki T, Kita N, Kanai M, Ashida T, et al. Levels of oxidative stress and redox-related molecules in the placenta in preeclampsia and fetal growth restriction. Virchows Arch 2004;444:49-55.
- Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA 1997;94:4028-33.
- Lin JH. Drug–drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev 2003;55:53-81.
- Novotna M, Libra A, Kopecky M, Pavek P, Fendrich Z, Semecky V, et al. P-glycoprotein expression and distribution in the rat placenta during pregnancy. Reprod Toxicol 2004;18:785-92.
- Lee CH. Induction of P-glycoprotein mRNA transcripts by cycloheximide in animal tissues: Evidence that class I Pgp is transcriptionally regulated whereas class II Pgp is posttranscriptionally regulated. Mol Cell Biochem 2001;216:103-10.
- Lee CH. Differential regulation of P-glycoprotein genes in primary rat hepatocytes by collagen sandwich and drugs. J Cell Biochem 2002;86:12-20.
- Gottesman MM. How cancer cells evade chemotherapy: sixteenth Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res 1993;53:747-54.
- Doi AM, Holmes E, Kleinow KM. P-glycoprotein in the catfish intestine: inducibility by xenobiotic and functional properties. Aquat Toxicol 2001;55:157-70.