
Sonic hedgehog protein promotes bone marrow-derived endothelial progenitor cell proliferation, migration and VEGF production via PI 3-kinase/Akt signaling pathways1
Introduction
Emerging evidence suggests that bone marrow contains postnatal endothelial progenitor cells (EPC), which contribute to tissue vascularization during postnatal physiological processes[1,2]. Recent preclinical and pioneering clinical studies have shown that the introduction of bone marrow-derived endothelial progenitor cells (BM-EPC) can restore tissue vascularization after ischemic events in limbs, retina and the myocardium[3,4]. Corecruitment of angiocompetent hematopoietic cells delivering specific angiogenic factors facilitates incorporation of EPC into newly sprouting blood vessels. Identification of cellular mediators and factors that promote proliferation and migration of EPC will open up new avenues of research to accelerate organ vascularization[5–8].
In the past decade, there has been increasing appreciation of the fact that pathways studied predominantly during embryogenesis and relatively silent during normal adult life, may be recruited postnatally in response to tissue injury[9]. Sonic hedgehog (Shh) proteins are morphogens that act in a wide variety of tissues during embryonic development and regulate epithelial-mesenchymal interactions that are crucial to morphogenesis of the nervous system, somite, limb, lung, gut, hair follicle, and bone[10]. Recently, it was reported that the shh pathway was involved in de novo vascularization of certain embryonic tissues as well as in inducing angiogenesis in an adult mammalian system[11]. But whether the shh signal pathway plays a role in regulating EPC behavior has not yet been proven. Hence, the purpose of the present study was to address the question of whether shh protein affected BM-EPC proliferation and migration, and then to further investigate the signaling mechanisms involved.
Materials and methods
Experimental reagents All cell culture reagents were obtained from Invitrogen (Carlsbad, CA, USA), including Medium 199, trypsin and fetal bovine serum (FBS). Rabbit anti-PI3-kinase antibodies were from Santa Cruz Biotechnology Inc(Santa Cruz, CA, USA). Enhanced chemiluminescent substrate (SuperSignal West Pico Chemiluminescent Substrate) was purchased from Pierce Chemical (Rockford, IL, USA). Hybrisol solution was purchased from Intergen (Purchase, NY, USA). Polyvinylidene difluoride and nylon membranes were purchased from Schleicher & Schüll (Dassel, Germany). Recombinant mouse sonic hedgehog N-terminus (shh-N), anti-mouse VEGF-neutralizing antibody (mVEGF-Ab) monoclonal, anti-human/mouse shh N-terminal peptide antibody were purchased from R&D Systems (Minneapolis, MN, USA). VEGF was from Collaborative Research (Bedford, MA, USA). Mouse VEGF ELISA Kits were purchased from Biocompare (South San Francisco, CA, USA). Rat anti-prominin-1 (Flk-1) R-phycoerythrin conjugated monoclonal antibody was from Chemicon (Temecula, CA, USA). Matrigel was purchased from B&D Biosciences (Heidelberg, Ger-many). Mouse VEGF, mouse basic fibroblast growth factor (bFGF), and human epidermal growth factor were purchased from Peprotech Inc (Rocky Hill, NJ, USA). H-7, LY294002 were purchased from Calbiochem (San Diego, CA). All other chemicals and reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise specified.
Animals Kunming mice, were purchased from the Center of Animals, Wuhan Academy of Preventive Medical Sciences. All animal studies were approved by the Animal Research Committee of Tongji College and were done according to the guidelines of National Institutes of Health (NIH). Congenic mice were bred and maintained at the animal research center of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Cell separation The murine bone marrow-derived mononuclear cells (MNC) were obtained from adult Kunming mice (4–6 weeks). Mice were killed by decapitation. Femora and tibiae were aseptically harvested, and the whole bone marrow was flushed using DMEM in a 1mL syringe with a 25-gauge needle. MNC were collected and washed twice with PBS. Pellets were resuspended in 5 mL of PBS supplemented with 0.5% bovine serum albumin (PBS buffer). Cells were manually counted by using a hemacytometer.
Cell purification Flk-1+cells were enriched using the MACS system (Miltenyi Biotec). Briefly, MNC were washed and resuspended in PBS buffer. The monoclonal antibody of anti-Flk-1 monoclonal antibody was linked with magnetic microbeads by SPA. Cells were first incubated with antibody labeled with MACS microbeads in a refrigerator at 6–10 ºC for 30 min. Labeled cells were loaded onto a column installed in a magnetic field. The column was rinsed with PBS buffer and negative cells passed through. Trapped cells were eluted after the removal of the column from the magnet. When a higher purity was needed, Flk-1+ positive fraction was reloaded onto a new column again. The column was rinsed with PBS buffer, positive fraction was eluted as described above.
BM-EPC culture BM-EPC were cultured according to previously described techniques[12]. Briefly, Flk-1 positive cells were plated on culture dishes coated with gelatin (Chemicon) and maintained in Medium 199 supplemented with 20% fetal-calf serum, VEGF (10 µg/L), bFGF (5 µg/L), EGF (5 µg/L). After 4 d culture, nonadherent cells were removed by washing with PBS, new media was applied, and the culture was maintained for 7 d and as were BM-EPC colony-forming units. The elongating and spindle-shaped cells were digested by trypsin and collected to use as BM-EPC.
Phenotypic analysis of BM-EPC-derived endothelial cells (EC) in vitro BM-EPC-derived EC were analyzed after 21 d of in vitro culture on gelatin-coated dishes in Medium 199 supplemented with 20% fetal-calf serum, which contains growth factors such as VEGF, bFGF, EGF, which are known to influence EC differentiation. Confirmation of endothelial phenotype was performed using endothelial-specific von Willebrand factor (vWF), and CD31, as previously reported. For detection of the vWF an anti-human vWF rabbit poly-clonal antibody (DAKO) that cross-reacts with murine antigen was used. Cells were fixed with 2% formaldehyde for 30 min, permeabilized with 0.2% saponin, incubated at 37 °C with the anti-human vWF antibody (3 mg/mL) for 1 h, and then incubated with 1.5 mg/mL Cy3-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, USA)
Effects of shh N-terminal peptide on BM-EPC proliferation The effects of shh N-terminal peptide on BM-EPC proliferation were evaluated using the MTT colorimetric assay. BM-EPC were seeded in triplicate 24-well plates (1×104 cells/well), and stimulated with different concentrations of shh N-terminal peptide (0.01, 0.1, 1, 10 µg/mL) at 0 h and again every other day, or VEGF (1 µg/mL) was used as a positive control. The cells were washed with PBS, trypsinized, and counted on d 0, d 1, d 2, d 3, d 4, and d 5. For the MTT assays, BM-EPC were seeded in triplicate 96-well plates (1×104 cells/well) and treated in an identical manner. Three days after treatment with shh N-terminal peptide, viable cell numbers were estimated by the MTT assay as described in a previous study[12]. Briefly, medium was removed and replaced with medium containing 5 mg/mL MTT and incubated for 4 h. The medium was then aspirated, and the product was solubilized with dimethyl sulfoxide. Absorbance was measured at 570 nm for each well using a microplate reader (Bio-Tek Instruments, Winooski, VT, USA) according to the manu-facturer’s protocol.
Flow cytometry analysis To further examine the proliferation-stimulating potential of shh N-terminal peptide in BM-EPC, we analyzed cell-cycle distribution after treatment with 1 μg/mL shh N-terminal peptide. Cells (1.5×106 cells) were cultured as described above, and 2 d later, they were fixed with 70% ethanol and incubated with 4 mmol/L phosphate-citrate buffer (4 mmol/L citric acid and 192 mmol/L Na2HPO4) for 30 min at room temperature. After centrifugation, cell pellets were resuspended in PBS containing propidium iodide/RNase (10 µg/mL each) and incubated for 20 min at room temperature. After the cell suspension was filtered with nylon film to eliminate any cell clumps, the cell cycle states were determined with a FACS Calibur (Becton-Dickinson Inc, San Jose CA, USA).
Cell migration Cell migration was assayed using a modified Boyden chamber technique (Transwell analysis). Porous filters (8 µm pores) were coated with type IV collagen on the both sides via passive absorption by incubating with 10 μg/mL collagen in coating buffer for 24 h. Serum-free medium containing different concentrations of shh N-terminal peptide (0.01, 0.1, 1, 10 µg/mL) was added into the lower chamber as a chemoattractant, and cells (1×104) were plated in the upper chamber and allowed to migrate for 24 h. Non-migrating cells were removed from the upper chamber with a cotton swab, filters were stained with Diff-Quik stain, and migrating cells adherent to the underside of the filter were enumerated using an ocular micrometer and by counting a minimum of 10 high-powered fields (HPF). Data are presented as relative migration (number of cells/HPF) and represent mean±SD of quadruplicate experiments.
Immunofluorescence Newly separated Flk-1 positive cells were washed twice with PBS and fixed with 4% paraformaldehyde for 10 min. Specimens were immunostained for the expression of VEGF. The process for immunostaining was performed as follows: fixed specimens were washed twice with PBS, then penetrated with 0.1% Triton-100 for 3 min, and washed twice with PBS. After being blocked with 4% calf serum for 20 min, specimens were incubated with the primary antibody against VEGF (1:200). Specimens were then washed with PBS, incubated with a fluorescein isothio-cyanate (FITC)-conjugated secondary antibody (1:500) at 37 ºC for 30 min, and washed with PBS and observed for positive signals employing fluorescence microscopy.
Enzyme-linked immunosorbent assay The production of VEGF in early BM-EPC was determined by ELISA as previously described in a previous study[23]. In brief, samples were added in duplicate to individual wells and incubated for 1 h at room temperature. After the plate was washed three times, 50 µL of biotin-conjugated VEGF antibody (4 µg/mL IgG) was added to each well. After incubation for 1 h at room temperature, streptavidin-HRP-conjugated goat antirabbit IgG antibody was added to individual wells, which were then incubated for 1 h at room temperature. About 50 µL of substrate containing o-phenylenediamine and hydrogen peroxide in citrate-phosphate buffer (pH 5.0) was added to each well. After incubation for 20 min at room temperature, the reaction was stopped with 4 mol/L sulfuric acid (25 µL). The absorbance at 492 nm was measured using an ELISA plate reader (Model 3550; Bio-Rad, CA, USA).
Protein extraction and Western blotting BM-EPC protein was extracted as described previously[13]. Briefly, the media in the 6-well plate was discarded, and cells were gently washed three times with cooled PBS. Lysis buffer (500 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 0.02% sodium azide, 0.1% SDS, 100 µg/mL phenylmethylsulfonyl fluoride, 1 µg/mL aprotinin, 1% Nonidet P-40, and 0.5% sodium deoxycholate) was added to the cells (0.25 mL/well). After incubation on ice for 30 min, the lysate was centrifuged at 12 000×g at 4 °C for 10 min. The protein concentration of the supernatant was determined using the Bradford method. Lysates (25 µg protein/lane) were resolved by SDS-polyacrylamide gel electrophoresis (12%), transferred to nitrocellulose membranes, and blocked with 5% nonfat dry milk in 10 mmol/L Tris-HCl, pH 7.5, 100 mmol/L NaCl, and 0.1% Tween 20. The membranes were then incubated with the primary rabbit antibody (1:150 dilution) overnight at 4 °C, followed by peroxidase-conjugated secondary antibody for 2 to 3 h. The ECL system was used to visualize the separated proteins. Blots were stripped and reprobed with β-actin as a loading control.
Evaluation of signaling pathways To examine the signaling mechanisms through which shh N-terminal peptide enhanced endothelial cell proliferation, migration, and angio-genesis, inhibitors of PI3-kinase (LY294002), PKC (H-7), anti-mouse VEGF-neutralizing antibody (mVEGF-Ab, 10 µg/mL) were added to cultured BM-EPC, and their effects on proliferation, migration, cell cycle and VEGF production were observed. As recommended by the manufacturers, inhibitors were used at concentrations of approximately 5× and 10× the lowest in vitro measured Ki, and their effects were reversible and not toxic at the concentrations used here. Stock solutions of protein kinase inhibitors were prepared as described in a previous study[14], and diluted into culture medium to obtain the desired final concentrations (12 µmol/L for H-7; 15 µmol/L for LY294002).
Statistical analysis Data were expressed as mean±SD. The data from FACS were evaluated by t-test.
Results
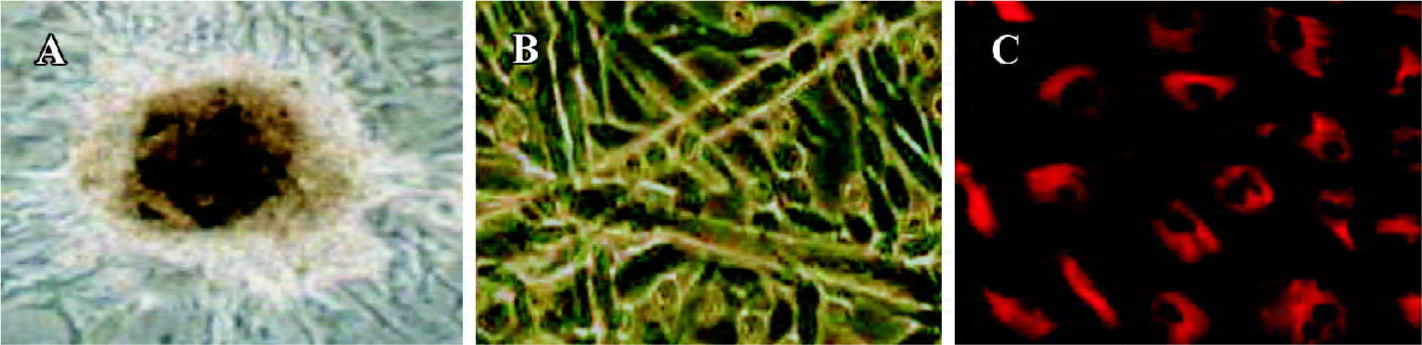
BM-EPC culture and phenotypic analysis The characteristics of endothelial cell progenitors were assessed by culture in Matrigel (gelatin), an extracellular matrix that can be used to promote differentiation of endothelial cells. As shown in Figure 1A, after 7 d in culture, BM-EPC colony forming units, defined as a central core of rounded cells surrounded by elongating and spindle-shaped cells, could be observed. Further cultured in Matrigel, the cells gradually lose their round appearance and assume the characteristic cobblestone morphology of mature endothelial cells (Figure 1B). Expression of endothelium-specific vWF was detected in rod-shaped granules that appear to be Weibel-Palade bodies (Figure 1C). These results provide additional support for the notion that the round cells represent endothelial cell progenitors.
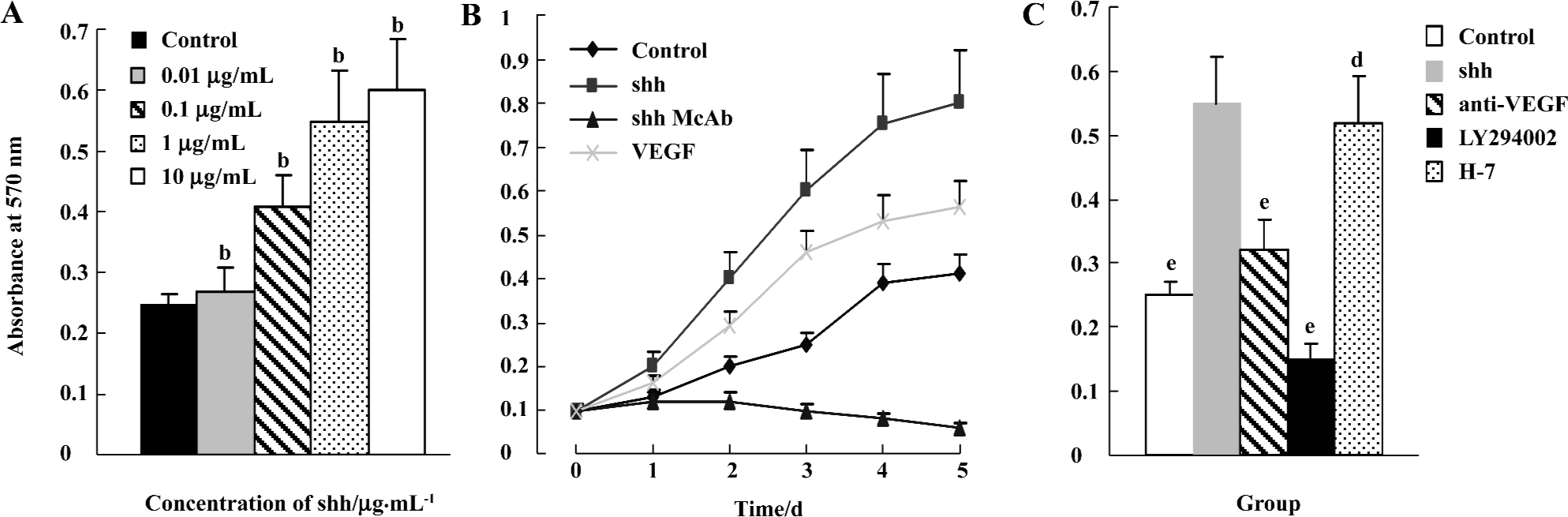
Effects of shh N-terminal peptide on BM-EPC proliferation To determine the effects of shh N-terminal peptide on BM-EPC proliferation, a necessary process for angiogenesis, we conducted experiments involving the direct addition of endogenous shh N-terminal peptide and then determined proliferative effects using MTT viability assays. The addition of individual shh N-terminal peptide significantly promoted proliferation of BM-EPC, and this effect was significantly inhibited by anti-human/mouse shh N-terminal peptide antibody (10 µg/mL). Furthermore, the effects of shh N-terminal peptide occurred at concentrations of 0.1 µg/mL up to10 µg/mL (Figure 2), lending support to the concept that shh N-terminal peptide promotes the proliferation of BM-EPC.
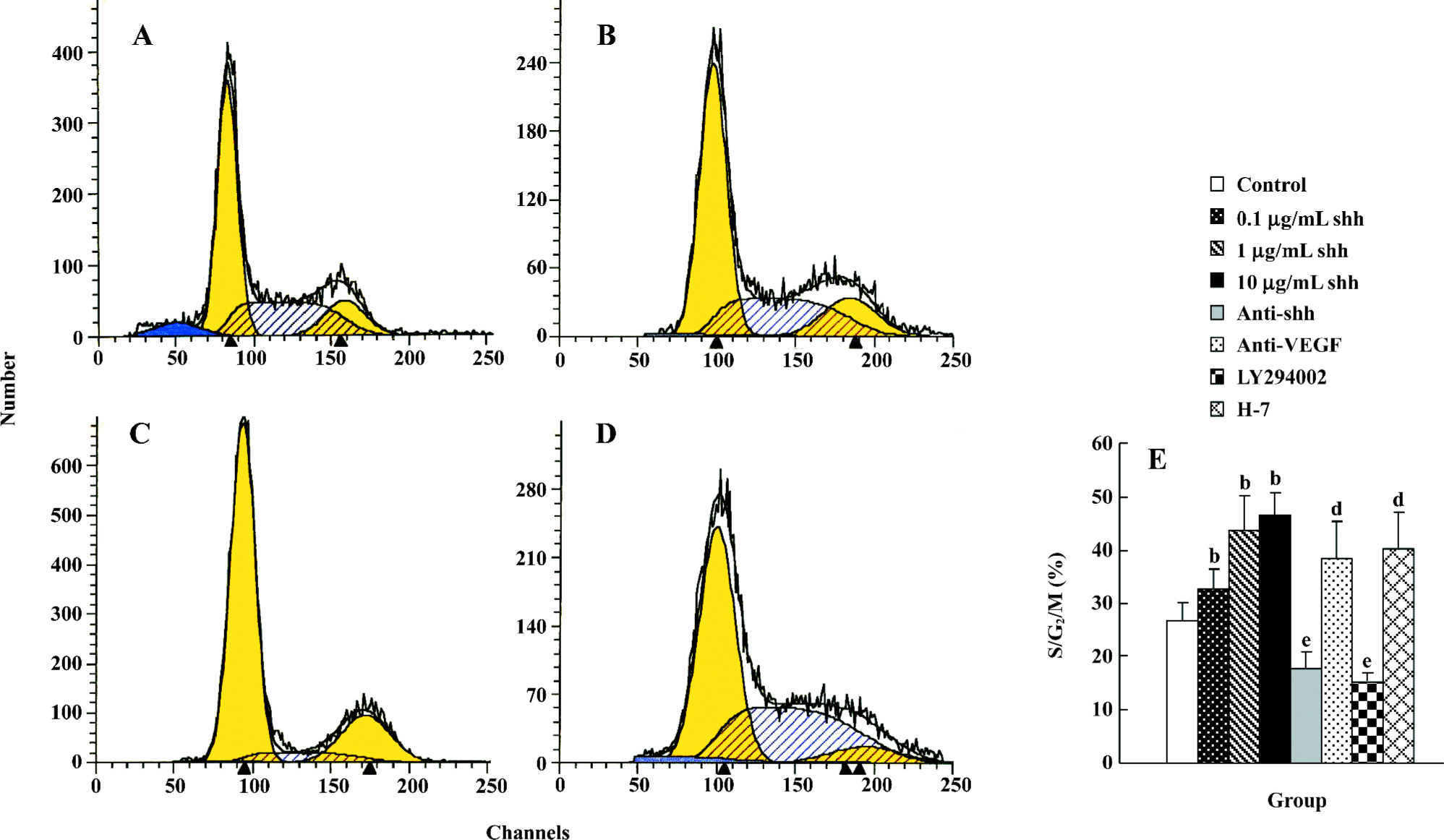
Flow cytometry analysis Cell cycle analysis by flow cytometry was performed after treatment with shh N-terminal peptide for 2 d. Treatment with 1 µg/mL of shh N-terminal peptide resulted in a significant increase in the proportion of cells in S/G2/M phase (43.67±0.65) compared with non-treated cells (26.83±1.65; P<0.01) (Figure 3A, 3B, 3E), and the effect stimulated with 1 µg/mL shh was significantly inhibited by anti-human/mouse shh N-terminal peptide antibody (10 µg/mL) (Figure 3C, 3E) or the inhibitor of PI3-kinase(LY294002, 15 µmol/L) (Figure 3E), but not by the inhibitor of PKC (H-7, 12 µmol/L) (Figure 3E). Furthermore, anti-VEGF partly inhibited the shh-mediated proliferation (Figure 3E). These data provide further evidence that shh N-terminal peptide promotes proliferation of BM-EPC, and suggest that shh promotes M-EPC proliferation via PI3-kinase, but not via the PKC pathway or through inducing VEGF production.
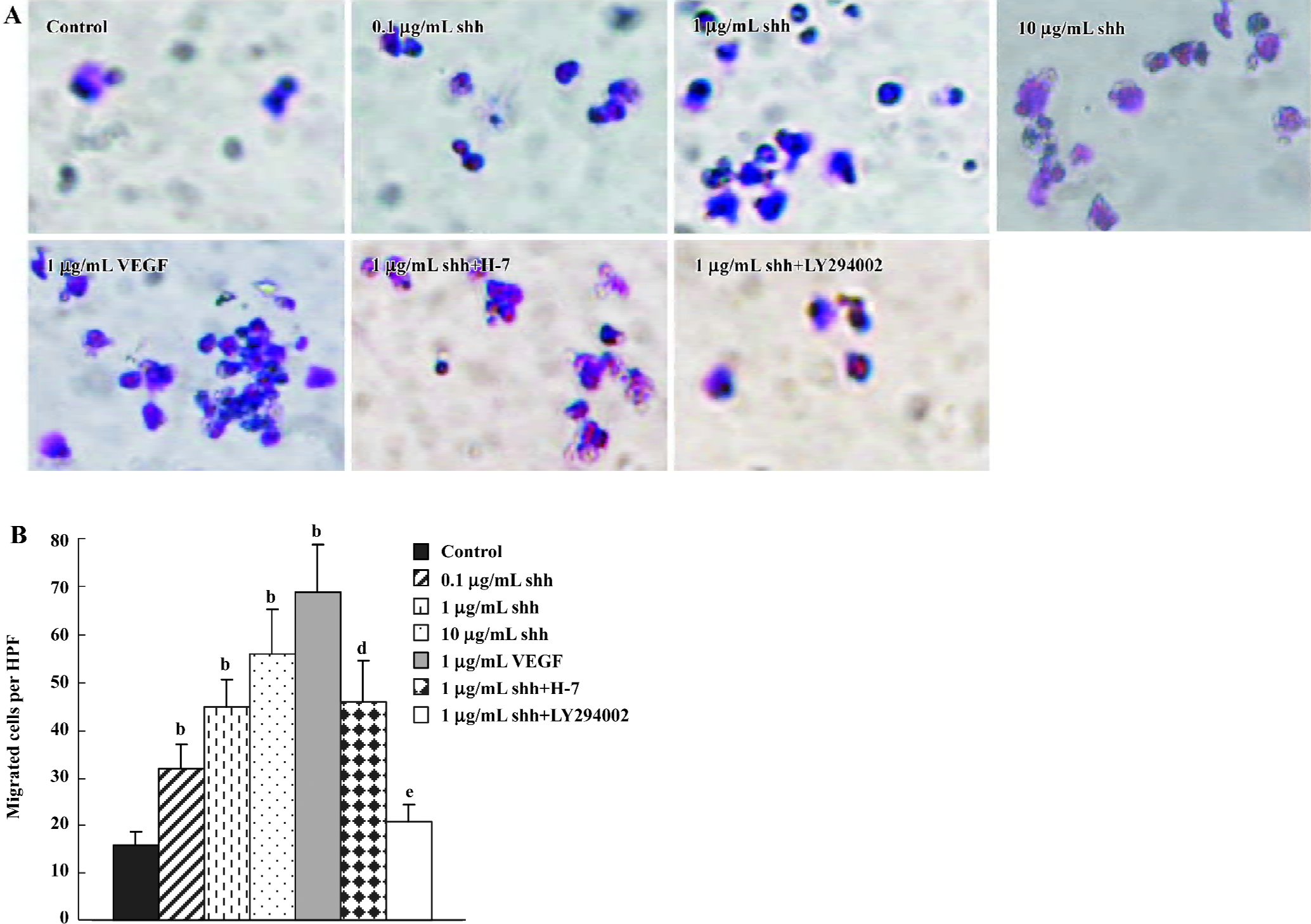
Cell migration In Transwell Boyden chamber assays (Figure 4), shh N-terminal peptide at concentrations of 0.1 µg/mL to10 µg/mL increased cell migration, and this effect was significantly inhibited by anti-human/mouse shh N-terminal peptide antibody (10 µg/mL). An addition of VEGF also promoted the migration of BM-EPC in Boyden chamber assays. Similar to their effects on shh-induced BM-EPC proliferation, inhibitors of the PI3-kinase (LY294002) markedly attenuated the shh-stimulated migration of BM-EPC, whereas the PKC inhibitor H-7 did not have an effect on the shh-mediated migration. These data suggest that PI3-kinase/Akt pathways play important roles in the shh-stimulated migration of BM-EPC.
Production of VEGF The newly separated Flk-1 positive cells produced low levels of VEGF, and when treated with shh N-terminal peptide at concentrations of 0.1 µg/mL to 10 µg/mL, the VEGF production was significantly increased (Figure 5A). The effect of shh in inducing VEGF could be attenuated by the inhibitor of PI3-kinase. Using an ELISA for mouse VEGF, we determined whether VEGF was present in supernatants from cultured BM-EPC. As shown in Figure 5B, VEGF is also present in the supernatants of cultured BM-EPC, but kept a very low level. And the level of VEGF in the supernatants of early BM-EPC was enhanced by shh N-terminal peptide at concentrations of 0.1 µg/mL to10 µg/mL, and this effect was significantly inhibited by anti-human/mouse shh N-terminal peptide antibody (10 µg/mL) or the inhibitors of PI3-kinase (LY294002), but could not be inhibited by PKC inhibitor (H-7).
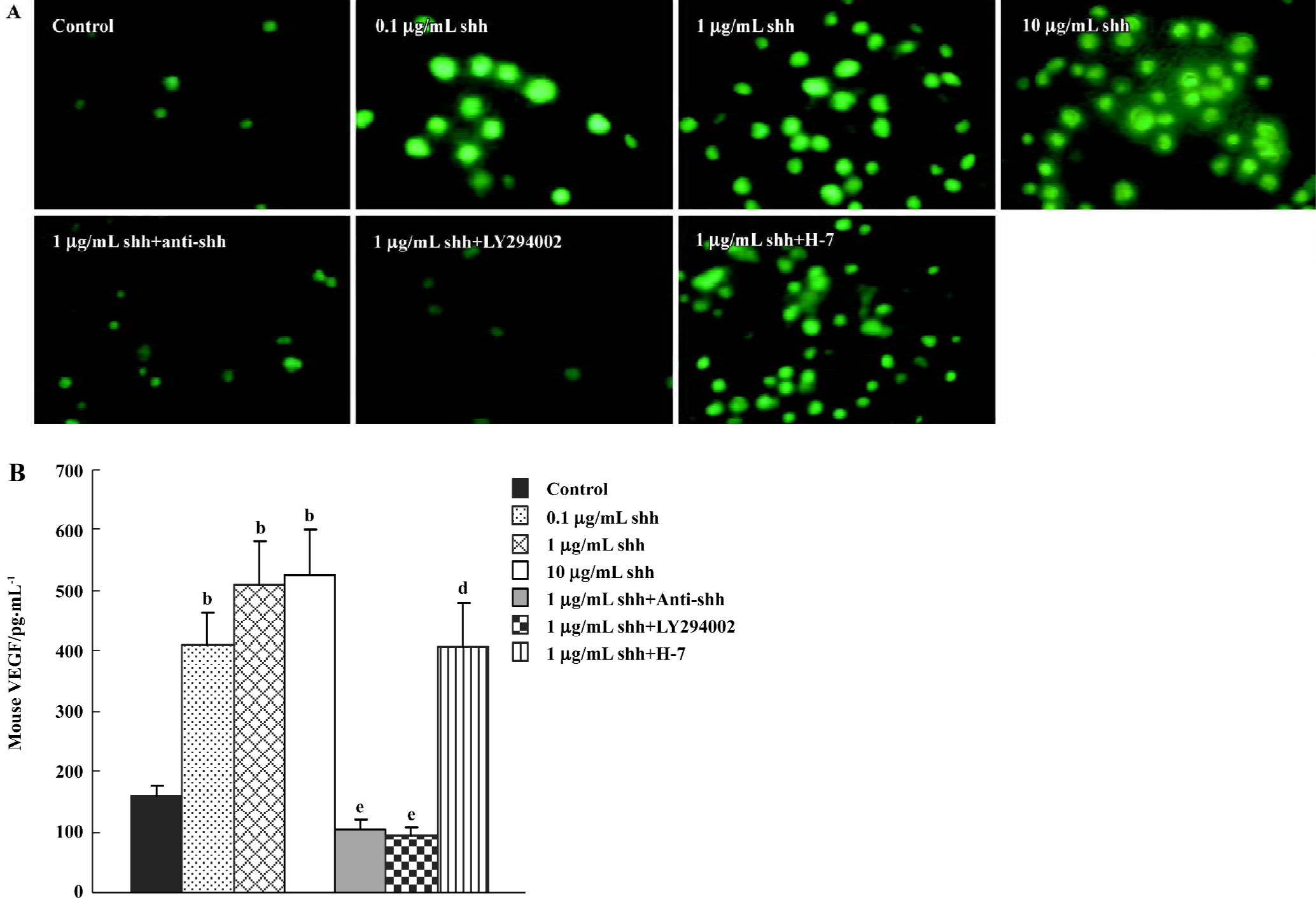
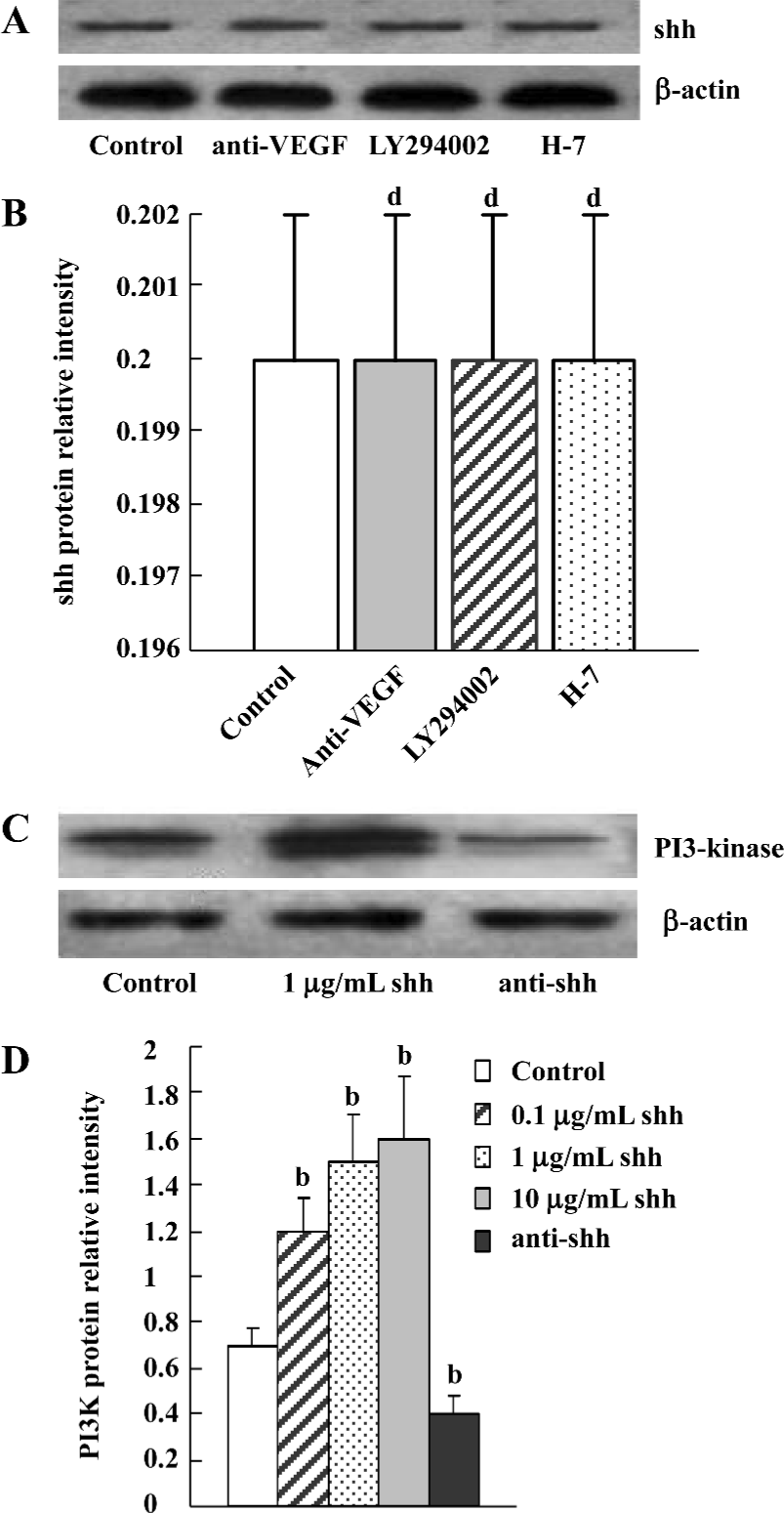
Evaluation of signaling pathways The newly separated BM-EPC could produce low levels of shh protein, and the production of shh protein could not be inhibited by the inhibitor of PI3 kinase, VEGF antibody or the inhibitor of PKC (Figure 6A, 6B). At the same time, there was low levels of phospho-PI3-kinase in BM-EPC, and the expression of phospho-PI3 kinase could be promoted by exogenous shh N-terminal peptide at concentrations of 0.1 µg/mL to 10 µg/mL, the level of phospho-PI3-kinase could be attenuated by anti-human/mouse shh N-terminal peptide antibody (Figure 6C, 6D), suggesting the PI3-kinase/Akt pathway is one of the downstream substrates of the shh signal pathway. And MTT assays of proliferation also showed shh promoted BM-EPC proliferation via PI3-kinase, but not via the PKC pathway (Figure 1C).
Discussion
Rapid revascularization of injured, ischemic and regenerating organs is essential to restore organ function. The angiogenic switch initiates the revascularization process and involves recruitment of endothelial cells that assemble into neovessels. Much effort has been focused on delivering angiogenic factors to accelerate tissue revascularization. However, because tissue injury is associated with the disruption of a permissive microenvironment necessary for recruiting pre-exiting EC, the exogenous introduction of vascular progenitor may facilitate restoration of organ revascularation[15,16]. Accumulating evidence suggests that bone marrow contains endothelial progenitor cells, a scarce population that contributes to the neoangiogenic processes. Though exceedingly rare, BM-EPC may function throughout a lifetime as a reservoir and/or a steady state source of primitive hematopoietic and endothelial cells[17]. But the mechanism of proliferation, recruitment, mobilization and incorporation of BM-EPC into neoangiogenic vessels remains unknown.
In a previous study, we proved that embryonic aorta-gonad-mesonephros (AGM)-derived stromal cell could promote proliferation of the production of blast-colony-forming cells (BL-CFC), which have the features of hemangioblast[18]. The role of shh in embryonic vascularization was discovered by gain- and loss-of-function studies in mice. Transgenic mice reveal that overexpression of shh in the dorsal neural tube results in hypervascularization of the neuroectoderm[19]. However, mice lacking shh function exhibit poor vascularization of the developing lung[20]. Recently, work in adult mice indicated that shh acted as an angiogenic factor, inducing robust neovascularization in hindlimbs damaged by ischemia, and that it could induce upregulation of proteins (VEGF-1, angiopoietins-1 and -2) required for the formation of blood vessels, and that it had the ability to upregulate these cytokines in concert so far seems unique to shh. Although, how shh upregulates angiogenic growth factors remains to be determined[21].
VEGF is an angiogenic factor that has been successfully used to treat severe myocardial ischemia and occlusive peripheral artery disease [22]. It also played a very important role in regulating mobilization of BM-EPC via inducing MMP-9 expression[23]. Considering the shh pathway is also a very important signal in regulating the proliferation and migration of stem cells[24], we speculated that shh might play a role in BM-EPC proliferation and migration by promoting VEGF production and so on.
The present study demonstrated that the addition of endogenous shh stimulated proliferation, migration and VEGF production in BM-EPC. Conversely, inhibiting shh activity by anti-shh antibodies leads to the inhibition of proliferation and migration of BM-EPC and a decrease of VEGF produc-tion, and demonstrated that shh could play a role in regulating the behavior of BM-EPC. Considering the effects of proliferation and migration mediated by shh were similar to the effects of VEGF, we next tried to determine whether the effects of shh observed in the present study were related to the upregulation of VEGF. We then examined the influence of anti-VEGF and found the addition of anti-VEGF to cultures of BM-EPC only partly attenuated the shh-induced migration and proliferation of these cells, suggesting that the shh pathway upregulation may be related, in part, to the angiogenic effects of VEGF. It seems likely that shh stimulated proliferation and migration of BM-EPC was not completely dependent on VEGF.
Some researchers recently reported that Sonic hedgehog induced capillary morphogenesis by endothelial cells through phosphoinositide 3-kinase, a well known pathway for regulating fundamental cellular responses such as proliferation, apoptosis, cell motility, and adhesion[25,26]. So in the present study, we tried to test the hypothesis that the PI3-kinase/Akt pathway is involved in proliferation, migration and VEGF production mediated by shh protein. We then observed the effects of the inhibitor of the PI3-kinase/Akt pathway on the shh-mediated proliferation, migration and VEGF production in BM-EPC in contrast with the inhibitor of PKC, another important pathway that was reported to regulate proliferation and migration of intact EC, and angiogenesis. It was observed that there was low levels of phospho-PI3-kinase in newly separated BM-EPC, but the expression of phospho-PI3 kinase could be promoted significantly by exogenous shh N-terminal peptide at concentrations of 0.1 µg/mL to 10 µg/mL. At the same time, the level of phospho-PI3-kinase could be attenuated by anti-human/mouse shh N-terminal peptide antibody, suggesting that the PI3-kinase/Akt pathway is one of the downstream substrates of the shh signal pathway. Moreover, it was demonstrated that the inhibitor of the PI3-kinase, but not the inhibitor of the PKC, almost completely inhibited the shh-mediated proliferation, migration and VEGF production, which was consistent with the concept that the PI3-kinase/Akt pathway may mediate the angiogenic effects of shh. Together, these data indicate that the shh-mediated effects on BM-EPC proliferation, migration and VEGF production likely occur via activation of PI3-kinase/Akt.
Our data demonstrate that shh protein promote an angiogenic phenotype, including BM-EPC proliferation and migration, and VEGF production. All of these processes are requisite steps in new vessel formation. The mechanisms underlying these effects may include activation of the PI3-kinase/Akt pathway, with VEGF production also contributing. Our observations also suggest the possibility that the shh protein may promote BM-EPC proliferation and migration to ischemic tissues. These results present the possibility of using shh for pro-angiogenic therapy in ischemic disorders.
References
- Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol 2004;287:C572-9.
- Fan CI, Gao PJ, Che ZQ, Liu JJ, Wei J, Zhu DL. Therapeutic neovascularization by autologous transplantation with expanded endothelial progenitor cells from peripheral blood into ischemic hind limbs. Acta Pharmacol Sin 2005;26:1069-75.
- Masuda H, Asahara T. Post-natal endothelial progenitor cells for neovascularization in tissue regeneration. Cardiovasc Res 2003;58:390-8.
- Aicher A, Zeiher AM, Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension 2005;45:321-5.
- Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, et al. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported” nitric oxide synthase activity. Circulation 2005;111:1114-20.
- Iwami Y, Masuda H, Asahara T. Endothelial progenitor cells: past, state of the art, and future. J Cell Mol Med 2004;8:488-97.
- Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Therapeutic potential of endothelial progenitor cells in cardiovascular diseases. Hypertension 2005;46:7-18.
- Hristov M, Weber C. Endothelial progenitor cells: characteriza-tion, pathophysiology, and possible clinical relevance. J Cell Mol Med 2004;8:498-508.
- Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, et al. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med 2005;11:1197-204.
- Gianakopoulos PJ, Skerjanc IS. Hedgehog signaling induces cardiomyogenesis in P19 cells. J Biol Chem 2005;280:21022-8.
- Pola R, Ling LE, Aprahamian TR, Barban E, Bosch-Marce M, Curry C, et al. Postnatal recapitulation of embryonic hedgehog pathway in response to skeletal muscle ischemia. Circulation 2003;108:479-85.
- Sladowski D, Steer SJ, Clothier RH, Balls M. An improved MTT assay. J Immunol Methods 1993;157:203-7.
- Fu JR, Liu WL, Zhou JF, Sun HY, Zheng M, Huang M. The effects of Mcl-1 gene in ATRA-resistant HL-60 cell. Chin J Heamatol 2005;26:352-4.
- Lin P, Welch EJ, Gao XP, Malik AB, Ye RD. Lysophosphatidyl-choline modulates neutrophil oxidant production through elevation of cyclic AMP. J Immunol 2005;174:2981-9.
- Gehling UM, Ergun S, Schumacher U, Wagener C, Pantel K, Otte M, et al. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood 2000;95:3106-12.
- Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med 2003;9:702-12.
- Park C, Ma YD, Choi K. Evidence for the hemangioblast. Exp Hematol 2005;33:965-70.
- Fu JR, Liu WL, Zhou YF, Zhou JF, Sun HY, Luo L, et al. Expansive effects of aorta-gonad-mesonephros (AGM)-derived stromal cells on HSCs from ESC. Chin Med J 2005;118:1979-86.
- Nagase T, Nagase M, Yoshimura K, Fujita T, Koshima I. Angiogenesis within the developing mouse neural tube is dependent on Sonic hedgehog signaling: possible roles of motor neurons. Genes Cells 2005;10:595-604.
- Miller LA, Wert SE, Clark JC, Xu Y, Perl AK, Whitsett JA. Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev Dyn 2004;231:57-71.
- Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake PR, et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med 2001;7:706-11.
- Kliche S, Waltenberger J. VEGF receptor signaling and endothelial function. IUBMB Life 2001;52:61-6.
- Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J 1999;18:3964-72.
- Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 2005;132:335-44.
- Kanda S, Mochizuki Y, Suematsu T, Miyata Y, Nomata K, Kanetake H. Sonic hedgehog induces capillary morphogenesis by endothelial cells through phosphoinositide 3-kinase. J Biol Chem 2003;278:8244-9.
- Sasaki T, Irie-Sasak J, Jones RG, Stanford WL, Bolon B, Wakeham A, et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 2000;287:1040-6.