
Expression and purification of lipoprotein-associated phospholipase A2, a key enzyme involved in atherosclerosis1
Introduction
Atherosclerosis is the major cause of many cardiovascular conditions and it is, primarily, a chronic inflammatory disease. Lipoprotein-associated phospholipase A2 (Lp-PLA2), also known as a platelet activating factor (PAF) acetylhydrolase, plays a key role in atherogenesis through the release of pro-inflammatory signals[1]. The Lp-PLA2 protein, which consists of 441 amino acids and has a predicted molecular weight of 45 kDa[2], is a member of the phospholipase A2 superfamily and belongs to the VII phospholipase A2 subgroup. The protein is produced mainly by monocytes, macrophages, T-lymphocytes, and mast cells, and can hydrolyze PAF, PAF-like phospholipids and polar-modified phosphatidylcholines[3]. The majority of Lp-PLA2 in human plasma (approximately 70%) is associated with low density lipoprotein (LDL), and the remainder is associated with high density lipoprotein (HDL). Lp-PLA2 can hydrolyze oxidized LDL to generate lysophosphatidylcholine (lyso-PC) and oxidized fatty acids; both of these hydrolysis products are pro-inflammatory factors that have pro-atherosclerotic effects. The increased levels of lyso-PC in oxidized LDL can be completely attributed to Lp-PLA2[3,4], the expression of which is regulated by inflammatory mediators. Lp-PLA2 activity is upregulated in atherosclerotic lesions, especially in complex plaques, and Lp-PLA2 appears to be an independent predictor of coronary artery disease (CAD) status[5,6]. It has been proposed that high plasma Lp-PLA2 levels should be viewed as a potential risk factor for CAD, and that Lp-PLA2 monitoring could provide information complementary to traditional lipoprotein measurements[7–9]. As such, Lp-PLA2 has been proposed as an important target molecule for the design of new treatment strategies for coronary heart disease (CHD)[10–12].
There is a clear need for inhibitors of Lp-PLA2 to more fully understand the role of this lipase in the atherosclerotic process. Use of Lp-PLA2 inhibitors is a new method separated from current lipid-lowering approaches for atherosclerosis therapy[13,14]. However, the quantity of Lp-PLA2 that can be purified from human serum is limited[15], so it is necessary to express this protein through a proper expression system for the study of Lp-PLA2 and its inhibitors. The identification of Lp-PLA2 inhibitors would aid our evaluation of the role of this enzyme in atherosclerosis, and the inhibitors can be potentially developed into new drugs for treating atherosclerosis. In the present paper, we report cloning of the Lp-PLA2 gene from THP-1 cells, and the expression and purification of Lp-PLA2. The recombinant Lp-PLA2 was enzymatically active and it could be used to establish a screening model for Lp-PLA2 inhibitors.
Materials and methods
Materials RPMI-1640 medium and Grace’s insect cell culture medium were purchased from Gibco (Carlsbad, USA). Sf-900 II serum-free medium was obtained from Invitrogen (Carlsbad, USA). Phorbol 12-myristate 13-acetate (PMA) was obtained from BioMol (Plymouth Meeting, USA). The Trizol reagent, Cellfectin reagent, and baculovirus expression vector system were purchased from Invitrogen (Carlsbad, USA). The M-MLV reverse transcriptase, restriction endonucleases, and T4-DNA ligase were obtained from Promega (Madison, USA). The oligonucleotides were synthesized by Sangon (Shanghai, China). The Pyrobest DNA polymerase was obtained from TaKaRa (Dalian, China). The Lp-PLA2 antibody was purchased from Cayman (Ann Arbor, USA). 1-O-alkyl-2[3H]acetyl-sn-glycero-3-phosphocholine [(3H-acetyl)PAF] was purchased from PerkinElmer (Wellesley, USA). The Ni-NTA His·Bind resin was obtained from Novagen (Nottingham, UK), and SB435495 was supplied by the Department of Medicinal Chemistry, Shanghai Institute of Materia Medica.
Gene cloning of Lp-PLA2 from THP-1 cells THP-1 cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). The cells were seeded in 6-well plates at a density of 8×108 cells/L and incubated for 6 h, and then the cells were stimulated to differentiate with the addition of 81 nmol/L PMA. Forty-eight hours later, the differentiated THP-1 cells were collected and total RNA was extracted using Trizol reagent. The total mRNA was reverse transcribed with 200 U M-MLV reverse transcriptase, 1 µL 0.5 g/L oligo d(T)18-primer and 0.5 mmol/L dNTPs, and the resulting cDNA was isolated and amplified by polymerase chain reaction (PCR) with Lp-PLA2-specific primers (sense, 5'-GCG C
Construction of donor vector The PCR product was electrophoresed in a 1.5% (w/v) agarose gel and purified with an agarose gel purification kit (Watson, Shanghai, China) in accordance with the manufacturer’s instructions. The purified DNA fragment was digested with Sal I and Xho I, and cloned into the Sal I- and Xho I-digested pFastBac1 donor vector. The nucleotide sequence of the fragment cloned into the plasmids was confirmed by DNA sequencing.
Generation of recombinant baculovirus transfer vector The validated pFastBac1 plasmids were amplified and isolated from DH5α. We used 0.1–1 µg of this DNA to transform 100 µL of competent DH10BAC cells. Colonies containing the recombinant bacmid were white, and colonies containing the unaltered bacmid were blue. The white colonies were selected for isolation of recombinant bacmid DNA. The white candidate colonies were streaked on isopropyl-1-thio-D-galactopyranoside (IPTG) plates to ensure that they were truly white colonies. Recombinant bacmid DNA was isolated from bacterial transformants using standard procedures with a minor modification (all samples were centrifuged at 18 000×g instead of 14 000×g). The recombinant bacmid DNA was verified to be a correct transcript by PCR.
Generation of recombinant baculovirus Sf9 cells were grown in Grace’s insect cell culture medium containing 10% FBS. The cells were transfected with recombinant bacmid using Cellfectin reagent as intermedia. Viral supernatant was collected at 72 h post-transfection and the viruses were amplified by infecting Sf9 cells with the viral supernatant. The recombinant viruses were subjected to 2 rounds of amplifications in order to obtain a high-titer stock. All virus stocks were stored at -20 °C and protected from light to ensure maintenance of titer.
Expression of recombinant Lp-PLA2 in insect cells Sf9 cells were grown in Grace’s insect cell culture medium supplemented with 10% FBS at first, and then were adapted to grow in Gibco Sf-900 II serum-free medium at 27 °C. To examine the time course of Lp-PLA2 expression, Sf9 cells (6×106/well) were seeded into a 6-well plate and grown in 2 mL SF900 II medium. The cells were infected with recombinant baculo-viruses at a multiplicity of infection (MOI) of 0.1. Cells were harvested at different time points by 5 min centrifugation at 500×g. For larger scale expression of (His)6-Lp-PLA2, Sf9 cells were cultured in 75 cm2 flasks and infected with recombinant high-titer virus stock. The cells were harvested at 48–96 h post-infection. Harvested cells were washed twice with 0.1 mol/L phosphate buffer (pH 7.4). The cells were resuspended in 8 volumes of lysis buffer (50 mmol/L Tris, 300 mmol/L NaCl, 10 mmol/L imidazole, 10 mmol/L CHAPS, 1 mg/L antipain, 10 mg/L leupeptin, and 1 mg/L pepstatin A; pH 7.5) and sonicated on ice for 80 s. The lysate was centrifuged at 18 000×g for 30 min at 4 °C and the clear supernatant was collected and kept at -80 °C until further processing.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting Proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a 5% polyacrylamide stacking gel and a 10% polyacrylamide separation gel. The proteins were visualized in the gels by staining with Coomassie brilliant blue R-250. For Western blot analysis, the proteins were transferred to nitrocellulose membranes and nonspecific binding was blocked with Tris-buffered saline (TBS; 100 mmol/L Tris-HCl, pH 7.5, 155 mmol/L NaCl) containing 3% albumin (bovine, fraction V; Amresco, Solon, USA). The nitrocellulose membrane was incubated with Lp-PLA2 antibody (1:1000) for 2 h at room temperature, and then with horseradish peroxidase-conjugated anti-rabbit IgG (Calbiochem, San Diego, USA) for 2 h at room temperature. The immune complexes were detected and visualized using the SuperSignal West Pico Trial Kit (Pierce, Rockford, USA) according to the manufacturer’s instructions.
Purification of recombinant Lp-PLA2 All purification assays were performed at 0 °C, unless stated otherwise. The thawed supernatant was recentrifuged at 18 000×g for 30 min (to ensure that no residual cell debris was present) and the clear supernatant was removed to a fresh tube. Saturated ammonium sulfate was added slowly into the supernatant to obtain a stepwise increase in ammonium sulfate concentra-tions. The solution was put on ice overnight to allow the precipitation process to conclude, and then centrifuged to collect the protein precipitate. The protein precipitate was dissolved in lysis buffer (50 mmol/L Tris, 300 mmol/L NaCl, 10 mmol/L imidazole, 10 mmol/L CHAPS, 1 mg/L antipain, 10 mg/L leupeptin, and 1 mg/L pepstatin A; pH 7.5). The solution containing the soluble His6-Lp-PLA2 was incubated with 1.5 mL 50% (v/v) Ni-NTA His·Bind resin (equilibrated in lysis buffer) at 4 °C with gentle rocking for 1 h. The mixture was then loaded on a 10 mL column under gravity flow and the column was washed twice with 6 mL wash buffer (50 mmol/L NaH2PO4, pH 8.0, 300 mmol/L NaCl, and 20 mmol/L imidazole). The (His)6-Lp-PLA2 was eluted from the column with 4×0.75 mL elution buffer (50 mmol/L Na H2PO4, pH 8.0, 300 mmol/L NaCl, and 250 mmol/L imidazole).
Enzyme assays Lp-PLA2 activity was measured using [3H]PAF as a substrate. All assays were performed at 37 °C in 50 mmol/L N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES) and 150 mmol/L NaCl, pH 7.4. Lp-PLA2 was incubated with 20 nmol/L [3H] PAF in a final volume of 200 µL for 10 min at 37 °C. The reaction was stopped by vortexing with 600 µL of CHCl3/MeOH (2:1), and the CHCl3 and aqueous layers were separated by a 15 min centrifugation at 12 080×g, and 250 µL of the upper aqueous layer was removed to a fresh tube and vortexed with 250 µL of CHCl3. The upper aqueous layer was again removed to a fresh tube and the amount of liberated [3H]acetate was determined by scintillation counting. The protein concentration of the fractions was measured using the Bradford’s method[16] with minor modifications. Briefly, 10 µL of protein solution was added into 3 mL of protein reagent and mixed by vortexing. The absorbance at 595 nm was measured after 10 min in 3 mL cuvettes against a protein reagent blank. The concentration of protein was determined according to a standard curve constructed using bovine serum albumin.
To examine the inhibitory effects of various compounds on the enzyme, purified Lp-PLA2 and the compounds were preincubated at 37 °C for 10 min before running the enzyme assay as described earlier.
Results
Gene cloning and generation of recombinant bacmid We used the THP-1 human acute monocytic leukemia cell line, and stimulated the cells to differentiate. The Lp-PLA2 gene was successfully cloned from the cDNA library of differentiated THP-1 cells, whereas Lp-PLA2 mRNA in non-stimulated THP-1 cells was not detected by PCR. We engineered the Lp-PLA2 gene to include a Kozak sequence for highly efficient translation initiation and a 6-His-tag sequence for purification. The fusion Lp-PLA2 gene was subcloned into the pFastBac donor plasmid. The amplified clones were sequenced in their entirety and found to be identical to the human sequence in the National Institutes of Health GenBank database (GenBank accession n
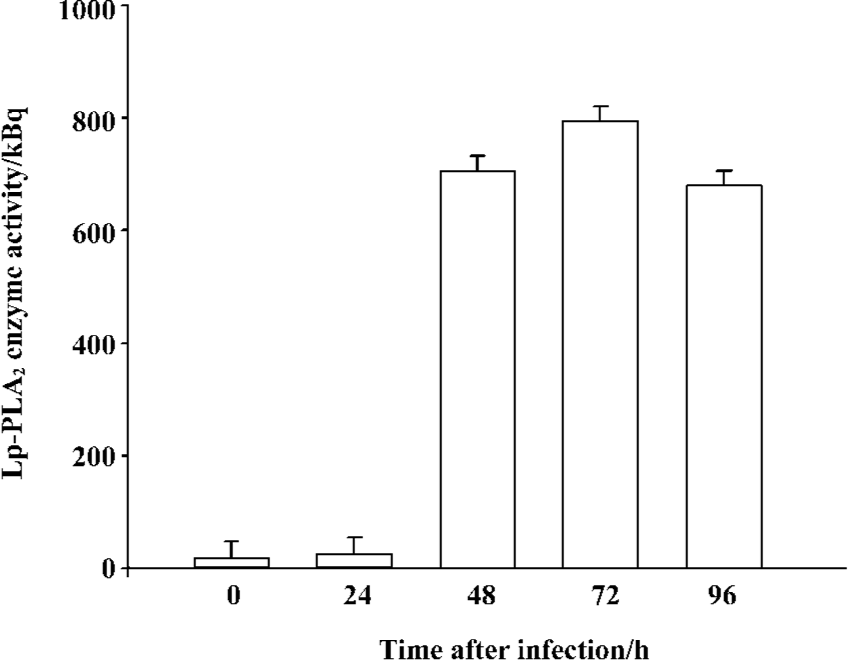
High-level expression of recombinant Lp-PLA2 in insect cells The recombinant bacmid was transfected into Sf9 cells and the baculovirus was amplified in the cells. The Sf9 cells were adapted in serum-free medium SF900-II, and the adapted Sf9 cells performed well in the SF900-II serum-free medium. We examined the effect of the Kozak sequence on the expression level of recombinant Lp-PLA2, and found that the Kozak sequence had greatly increased the expression level of Lp-PLA2. The recombinant Lp-PLA2 was expressed at a high level (Figure 1). As measured by densitometry scanning, the amount of recombinant Lp-PLA2 was up to 14.5% of the total cellular protein in the late stages of infection. No Lp-PLA2 was detected within the first 24 h post-infection (Figure 1B). To examine the time course of enzyme activity, cells were harvested at 0, 24, 48, 72, and 96 h after infection, and the enzyme activity of cell proteins was measured as described earlier. Our results revealed that the enzyme activity peaked 72 h after baculovirus infection (Figure 2).
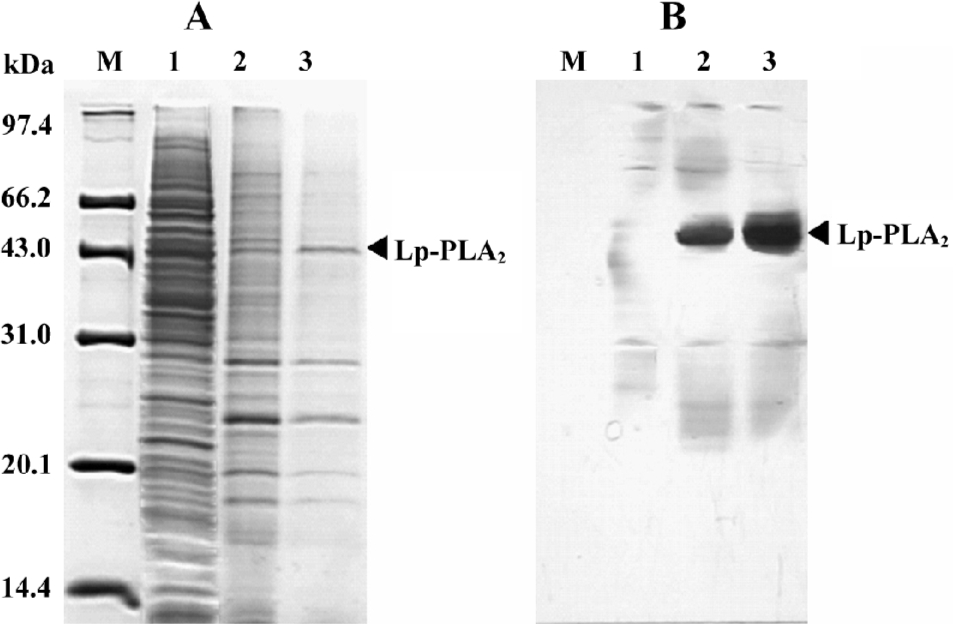
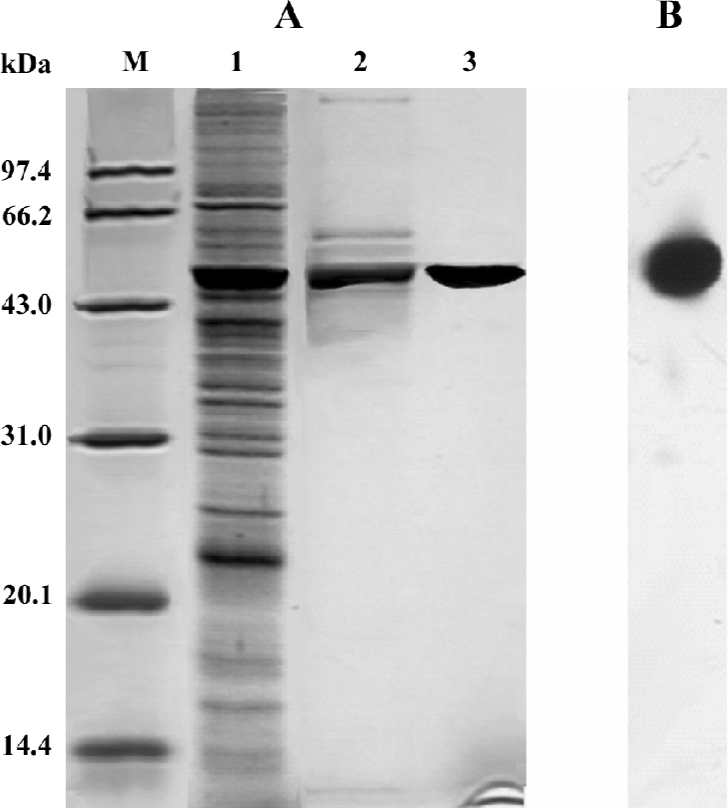
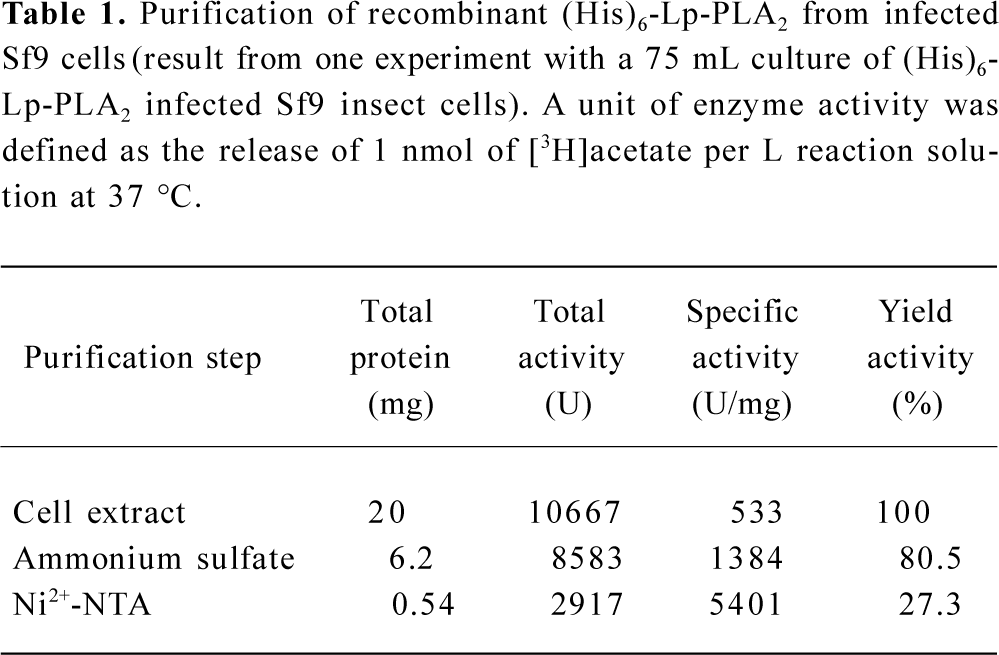
Purification of recombinant histidine-tagged Lp-PLA2 The cell extraction proteins were first subjected to a stepwise precipitation with different ammonium sulfate saturations. We found that most of the recombinant protein was precipitated, and that only a few other proteins precipitated when the ammonium sulfate was at 20% saturation. After the recombinant proteins were first partially purified by ammonium sulfate precipitation, the precipitate was redissolved in the lysis buffer and purified through Ni-NTA His·Bind resin. The histidine-tagged Lp-PLA2 protein was purified to near homogeneity after the purification process (Figure 3). The yield ratio was lower than 14.5% because some recombinant Lp-PLA2 was lost in the purification process. The final yield obtained from insect cells was 0.54 mg per 20 mg cell proteins with at least 95% purity, and the yield ratio was 2.7%, which was much greater than that obtained from human plasma. The final yield obtained from human plasma was 7.0 mg per 6440 mg plasma proteins, and the yield ratio was 0.11%. The results of a typical purification are summarized in Table 1.
Full table
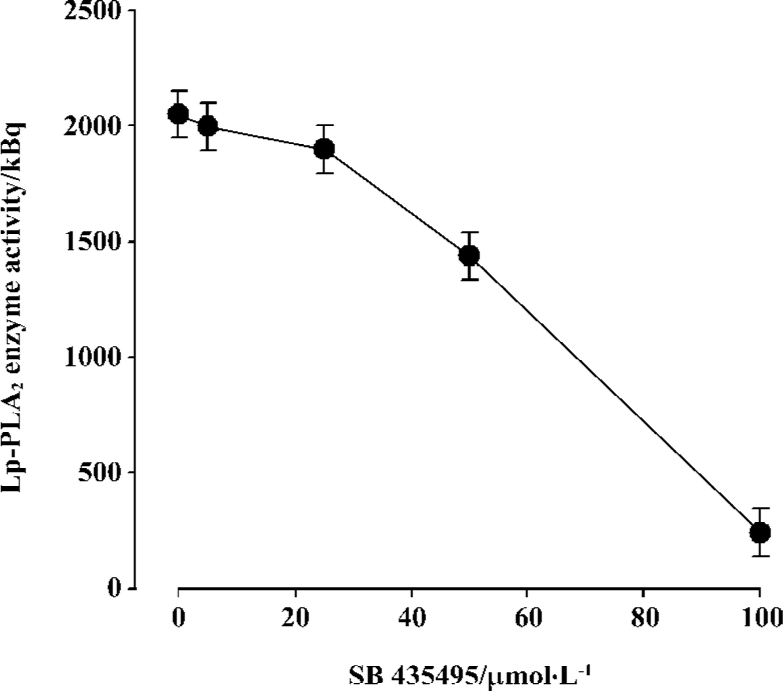
Inhibitive effect of SB435495 on recombinant Lp-PLA2 Recombinant Lp-PLA2 was first identified by Western blotting using the highly specific anti-Lp-PLA2 antibody (Figure 1B). The specific activity of (His)6-Lp-PLA2, using [3H]PAF as substrate, was measured (Table 1). In order to further identify and examine the usefulness of our recombinant Lp-PLA2 for screening Lp-PLA2 inhibitors, we examined the enzyme activity of recombinant Lp-PLA2 pretreated with the known Lp-PLA2 inhibitor SB435495. The inhibitor produced a good inhibitory curve for the inhibition of recombinant Lp-PLA2 (Figure 4) and it inhibited the recombinant enzyme with an IC50 of 57±1 µmol/L. These results collectively indicate that our recombinant Lp-PLA2 protein could be used to establish a screening model for Lp-PLA2 inhibitors, and that SB435495 could be used as a positive control.
Discussion
Lp-PLA2 is an inflammatory marker that has been identified as a novel therapy target for atherosclerosis[17–19]. Inhibition of Lp-PLA2 has anti-atherogenic effects. However, the large-scale screening of Lp-PLA2 inhibitors has proven to be difficult. Direct screening of plasma LDL may yield nonspecific inhibitors due to the presence of other phospholipase A2 molecules, while purification of Lp-PLA2 from plasma is complicated and produces a very low yield. Thus, we herein sought to develop a simple, large-scale method for producing recombinant Lp-PLA2 suitable for inhibitor studies.
In the present study, we reported the cDNA cloning of Lp-PLA2 from differentiated THP-1 cells. We engineered the N-terminal of the cDNA to possess a Kozak sequence for high-level translation initiation. We found that the Kozak sequence greatly increased the expression level of recombinant Lp-PLA2 in Sf9 insect cells. The amount of recombinant Lp-PLA2 was up to 8.5% of the total cellular protein, and the inclusion of the Kozak sequence further increased this expression up to 14.5% of total cellular proteins. A very high expression level in insect cells was achieved. We also adapted the Sf9 cells in serum free SF900-II media to remove the interference of serum in enzyme assays.
The recombinant Lp-PLA2 was constructed as a fusion protein with a 6-His tag at the C-terminus for convenient purification by affinity chromatography. The enzyme could not be purified directly using the Ni-NTA His·Bind resin at first, so we partially purified the protein by ammonium sulfate precipitation before the affinity purification step. The enzyme was efficiently purified through this 2-step procedure. The enzyme activity of the recombinant Lp-PLA2 was 115 µmol/min per mg, and it was at the same level of the activity of the enzyme purified from human plasma, which was 112 µmol/min per mg. The recombinant purified Lp-PLA2 was enzymatically active, and the inhibitor SB435495 inhibited the enzyme activity. Thus, using our recombinant Lp-PLA2, we could establish a screening system for Lp-PLA2 inhibitors.
In conclusion, we reported gene cloning of Lp-PLA2 from differentiated THP-1 cells. We demonstrated how to take advantage of recently developed molecular cloning technologies to successfully express and purify Lp-PLA2 at a high level in the insect cell-baculovirus expression system. The yield ratio was much greater than that obtained from human plasma. We established a screening model for Lp-PLA2 inhibitors, and the screening model would facilitate larger scale screening of Lp-PLA2 inhibitors and discovery of more novel inhibitors. The recombinant Lp-PLA2 and its novel inhibitors would aid further research regarding the role of this enzyme in atherosclerosis and the development of new drugs for atherosclerosis therapy.
Acknowledgements
We are grateful to Prof Xing-zu ZHU, Dr Wei-yu ZHANG, and Dr Wan-chun SUN for their helpful advice and technical support.
References
- Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol 2005;25:923-31.
- Tjoelker LW, Wilder C, Eberhardt C, Stafforini DM, Dietsch G, Schimpf B, et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995;374:549-53.
- Macphee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem J 1999;338:479-87.
- Carpenter KLH, Dennis IF, Challis IR, Osborn DP, Macphee CH, Leake DS, et al. Inhibition of lipoprotein-associated phospholipase A2 diminishes the death-inducing effects of oxidized LDL on human monocyte-macrophages. FEBS Lett 2001;505:357-63.
- Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR, Heiss G, et al. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation 2004;109:837-42.
- Oei HH, van der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MM, et al. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation 2005;111:570-5.
- Koenig W, Khuseyinova N, Lowel H, Trischler G, Meisinger C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: results from the 14-year follow-up of a large cohort from southern Germany. Circulation 2004;110:1903-8.
- Brilakis ES, McConnell JP, Lennon RJ, Elesber AA, Meyer JG, Berger PB. Association of lipoprotein-associated phospholipase A2 levels with coronary artery disease risk factors, angiographic coronary artery disease, and major adverse events at follow-up. Eur Heart J 2005;26:137-44.
- Iribarren C, Gross MD, Darbinian JA, Jacobs DR Jr, Sidney S, Loria CM. Association of lipoprotein-associated phospholipase A2 mass and activity with calcified coronary plaque in young adults: the CARDIA study. Arterioscler Thromb Vasc Biol 2005;25:216-21.
- Dada N, Kim NW, Wolfert RL. Lp-PLA2: an emerging biomarker of coronary heart disease. Expert Rev Mol Diagn 2002;2:17-22.
- Packard CJ, O’Reilly DSJ, Caslake MJ, McMahon AD, Ford I, Cooney J, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med 2000;343:1148-55.
- Macphee CH. Lipoprotein-associated phospholipase A2: a potential new risk factor for coronary artery disease and a therapeutic target. Curr Opin Pharmacol 2001;1:121-5.
- Macphee C, Benson GM, Shi Y, Zalewski A. Lipoprotein-associated phospholipase A2: a novel marker of cardiovascular risk and potential therapeutic target. Expert Opin Investig Drugs 2005;14:671-9.
- Leach CA, Hickey DM, Ife RJ, Macphee CH, Smith SA, Tew DG. Lipoprotein-associated PLA2 inhibition: a novel, non-lipid lowering strategy for atherosclerosis therapy. Farmaco 2001;56:45-50.
- Tew DG, Southan C, Rice SQ, Lawrence MP, Li H, Boyd HF, et al. Purification, properties, sequencing and cloning of a lipoprotein associated, serine dependent phospholipase which is involved in the oxidative modification of low density lipoproteins. Arterio-scler Thromb Vasc Biol 1996;16:591-9.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-54.
- Rader DJ. Inflammatory markers of coronary risk. N Engl J Med 2000;343:1179-82.
- Tselepis AD, Chapman JM. Inflammation, bioactive lipids and atherosclerosis: potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Athero-scler Suppl 2002;3:57-68.
- Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipidol 2005;16:442-6.