
Expression of proline-rich Akt-substrate PRAS40 in cell survival pathway and carcinogenesis1
Introduction
Chemoprevention is a logical and obvious strategy to help alleviate the effects of cancer[1,2]. Much effort has been focused on the discovery and development of new chemopreventive agents, especially agents targeted at mechanisms known to be involved in the process of carcinogenesis[3,4].
The PI3K-Akt signaling pathway regulates many normal cellular processes including cell proliferation, survival, growth and motility, which are critical for tumorigenesis. The role of the PI3K-Akt signaling pathway in oncogenesis has been extensively investigated and altered expression or mutation of many components of this pathway have been implicated in human cancer[5] . Akt functions as a major downstream target of phosphatilylinositol-3-kinase (PI3K), carrying out functions including stimulation of glucose uptake and cell growth as well as inhibition of apoptosis[6,7].
Kovacina et al used a combination of the 14-3-3 protein and anti-pAkt substrate antibodies to screen and isolate a substrate of Akt, the major 14-3-3 binding protein observed in cells after insulin treatment (a 40 kDa molecule)[8]. This protein contains a consensus Akt phosphorylation site (Thr-246) but not other recognizable motif. It is highly proline-rich, with 15% of its amino acids being proline (versus 5% for a typical protein), and these proline-rich regions are potential SH3 and/or WW domain binding partners. The protein has therefore been named PRAS40, which stands for the “proline-rich Akt substrate of 40 kDa”. Activation of inducible Akt alone was sufficient to stimulate PRAS40 phosphorylation, and phosphorylation of this protein was reduced in cells lacking Akt1 and Akt2. Thus PRAS40 is a novel substrate of Akt, the phosphorylation of which leads to binding of this protein to 14-3-3[8]. Saito et al demonstrated that the expression of PRAS40, PRAS40/Akt and PRAS40/14-3-3 increased in Nerve Growth Factor (NGF) treated mice but decreased with inhibition of PI3K and the NGF receptor after transient focal cerebral ischemia (tFCI) [9].
However, the critical role of PRAS40 in the biological processes of cell proliferation and carcinogenesis remains unknown. The aim of our study was to examine the expression of PRAS40 in the cell survival signaling pathway, tumor progression and its relationship with Akt and 14-3-3. Information from this study will be helpful for further functional studies of PRAS40.
Materials and methods
Plasmids and expression The constitutively active Akt plasmid HA-Akt-ΔpH and kinase mutation Akt plasmid HA-Akt-Km were the gift of Prof Dr Haian FU (Emory university, USA). After transfection into human embryonic kidney cell HEK293, the supernatant was boiled for 5 min in sodium dodecylsulfate (SDS) sample buffer and resolved using SDS-polyacrylamide gel electrophoresis (SDS-PAGE, 12.5%), The proteins were transferred to polyvinylidene difluoride (PVDF) membranes (BIO-RAD, CA, USA). The primary antibodies were 1:2000 dilution of mouse monoclonal antibody against HA-Prob, Western blots were performed with horseradish peroxidase-conjugate anti-mouse IgG. Cross-reacting materials were visualized using ECL Detection Reagents (Amersham Biosciences, UK).
Source of antibody Rabbit polyclonal antibody against phosphorylated PRAS40(T246), and mouse monoclonal antibody against PRAS40 were purchased from BioSource International (Camarillo, CA, USA); Rabbit polyclonal antibodies against phosphorylated Akt (S473), Akt, phosphorylated-Raf (S338) and Raf were purchased from Cell Signaling Technology (Beverly, MA, USA); Rabbit polyclonal antibody against 14-3-3 (Pan-14-3-3), mouse monoclonal antibody against tubulin and HA-Prob, goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP were purchased from Santa Gruz Biotechnology (CA, USA).
Reagents Ly294002/worthmannin (PI3K inhibitor) were purchased from Cell Signaling Technology; rapamycin (mTOR inhibitor) was purchased from Calbiochemistry (USA); hydrogen peroxide and U0126 (MEK inhibitor) were obtained from Sigma-Aldrich (St Louis, MO, USA).
Cell culture and DNA transfection HEK293, A549, HeLa and other cancer cells were provided by the Winship Cancer Institute (Atlanta, GA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI1640 (GIBCO Invitrogen, CA, USA) with 10% heat inactivated fetal bovine serum (FBS) and 5% CO2 at 37 oC. The confluent cells were separated and trypsinized with 0.05% trypsin-ethylenediamine tetraacetic acid (EDTA) to produce single cells. They were then seeded at 1×105 per cm2 and allowed to form subcultures. Cells were transfected with plasmids using the FuGENE6 reagent (Roche Applied Science, USA) according to the manufacturer’s instructions.
Western blot After the cells were transfected, harvested and lysed by in 1% NP-40 lysis buffer [0.15 mol/L NaCl, 0.01 mol/L N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 1% NP-40, 5 mmol/L sodium pyrophosphate, 5 mmol/L sodium fluoride, 2 mmol/L sodium orthovanadate, 10 mg/L aprotinin, 10 mg/L leupetin, 1 mmol/L phenylmethlsulfonyl fluoride (PMSF)] at 4 oC for 20 min, cell extracts were clarified by centrifugation, prepared in SDS sample buffer, boiled for 5 min, and resolved using SDS-PAGE (12.5%) for Western blot. The enzyme-linked immunoblotting procedures were performed essentially as described previously[10]. Corresponding secondary antibodies were used against each primary antibody: horseradish peroxidase-conjugated goat anti-mouse IgG was used for monoclonal antibodies and horseradish peroxidase-conjugated goat anti-rabbit IgG was used for polyclonal antibodies. Cross-reacting materials were visualized using ECL detection reagents.
Saito et al suggested that PRAS40 might play a critical role in the neuronal cell survival pathways mediated by NGF after cerebral ischemia[9]. In order to test which cell survival signal cascade PRAS40 is involved in, we used three key kinase inhibitors (Worthmannin/Ly294002, Uo126, rapamycin) and cell death or apoptosis induction factors (serum withdrawal, H2O2) to treat normal cell line HEK293 and cancer cell line A549 and HeLa. The expression of PRAS40, Akt, Raf and 14-3-3 were analyzed by Western blot.
Results
PRAS40 was mainly involved in the PI3K-Akt survival pathway After treatment with the PI3K inhibitor Worthmannin (2 µmol/L) or Ly294002 (25 µmol/L) in HEK293 for 2 h, Worthmannin and Ly294002 almost completely blocked phospho-PRAS40 and phospho-Akt activity, compared with total expression of PRAS40 and Akt. However, inhibition of Akt phosphorylation was also induced by serum withdrawal. Overexpression of constitutively active Akt (Akt-ΔpH) promoted the expression of phopho-PRAS40 in HEK293, compared with negative control Kinase mutation Akt (Akt-Km)(Figure 1). Therefore, the main pathway that PRAS40 is involved in is the PI3K-Akt pathway, and Akt is the upstream kinase of PRAS40 .
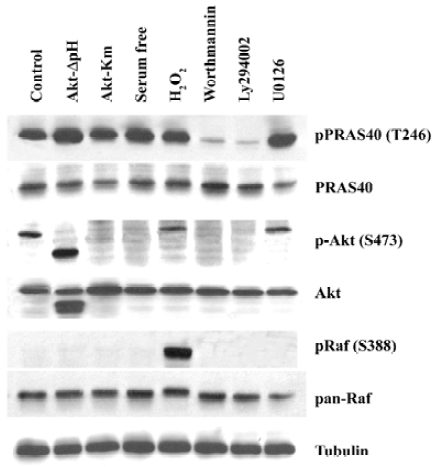
MEK-ERK pathway is not necessary for PRAS40 activity The Raf-dependent activity of the MEK-ERK pathway promotes cell survival by targeting various death pathways[11]. To test whether MEK-ERK is involved in the regulation of PRAS40/Akt activity, we used the MEK antagonist U0126 to treat HEK293, A549 and HeLa cell lines. Western blot analysis revealed that U0126 had no effect on PRAS40 activity (Figure 1, 2), which implies that PRAS40 does not contribute to the MEK-ERK pathway.
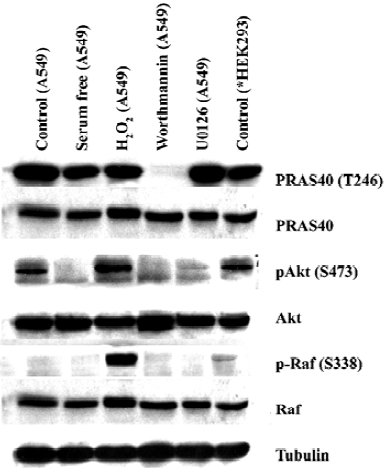
ROS induced phosphorylation of Raf/Akt activity but not PRAS40 at low concentration ASK1 is known to play an important role in the apoptotic response induced by ROS, in particular H2O2. Goldmann et al showed that phosphorylation of ASK1 at Ser-976 by H2O2 stimulation was dose-dependent, and that treatment with 1 mmol/L H2O2 for 30 min could completely dephosphorylate ASK1 at Ser-967[12]. In our experiment, the increase in phospho-Akt and phospho-Raf expression was induced by treatment with 1 mmol/L H2O2 for 20 min in HEK293 and A549, but there was no change on PRAS40 activity (Figure 1, 2).
Akt was not the only upstream kinase of PRAS40 Akt phosphorylation could be blocked by worthmannin, U0126 and serum withdrawal in A549 and HEK293 cells; however, only worthmannin and Ly294002 inhibited phospho-PRAS40 activity (Figure 1, 2), Therefore, Akt was not the only upstream kinase of PRAS40. PRAS40 could also be activated by an Akt-independent mechanism. Western blot analysis showed that the phospho-PRAS40 and phospho-Akt activity in A549 cancer cells were stronger than in HEK293 normal cell line (Figure 2).
Expression of PRAS40 and 14-3-3 was higher in HeLa than in HEK293 cells The 14-3-3 protein is an anchor protein for some Akt substrates, and it is also a PRAS40 binding protein. Therefore, we investigated the relationship between PRAS40 and 14-3-3 expression in normal and cancer cells. HEK293 and HeLa cells were treated with serum withdrawal, H2O2, rapamycin (mTOR inhibitor), worthmannin and U0126. Western blot demonstrated that, except for worthmannin, these treatments produced no significant difference with regard to PRAS40 activity in HEK293 and HeLa cells. We found that HEK293 was more sensitive: treatment with worthmannin only decreased the phosphorylation of PRAS40 in HeLa cells, but completely blocked PRAS40 phosphorylation in HEK293 cells. We also found that phospho-PRAS40 and 14-3-3 were expressed more strongly in HeLa cells than in HEK293 cells, and the total expression of PRAS40 was almost the same in the two cell lines (Figure 3).
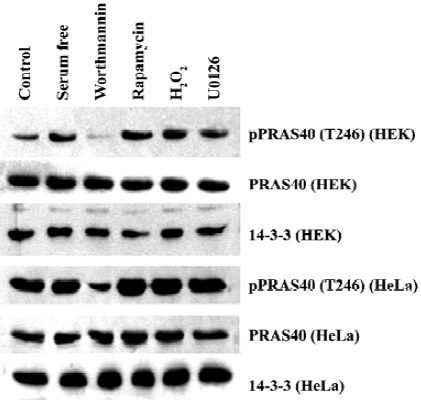
We also noticed that there was a difference in sensitivity to worthmannin between normal cells and cancer cells (HEK293 and HeLa) and among cancer cells (A549 and HeLa).
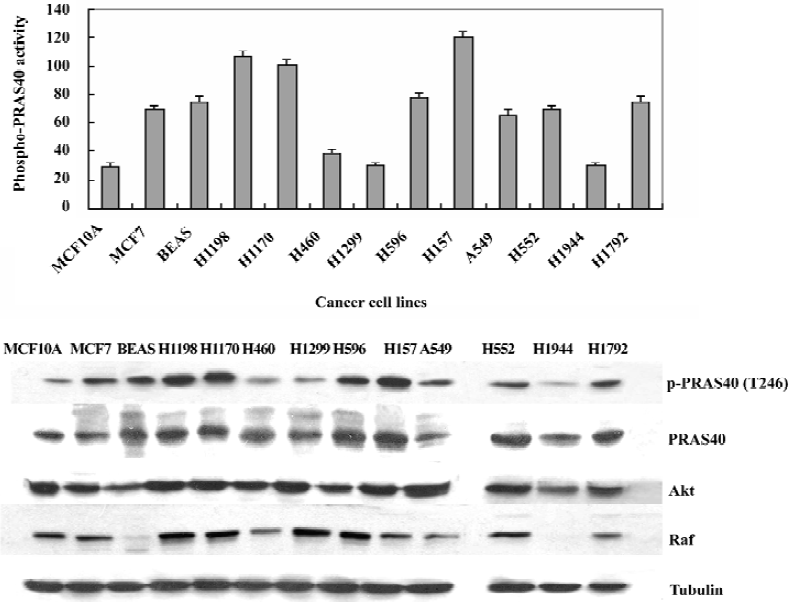
PRAS40 phosphorylation activity was stronger in cancer cells than in normal cells In order to determine if PRAS40 activity is different in normal cells, different types and different development phases of cancer cells, we used Western blot analysis to determine the expression of phospho-PRAS40, PRSA40, Akt and Raf in vitro. Because Akt and Raf are the key control factors in the cell survival pathway (PI3K-Akt pathway and Raf-MEK pathway), we used them to find a expression relationship between PRSA40, Akt and Raf.
For a breast cancer model, we used the MCF10A/MCF7 cell lines. MCF10A is a normal breast cell line and MCF7 is a breast carcinoma line. For a lung cancer model, we used the BEAS/H1190/H1170 cell lines. BEAS cells are Human bronchial epithelial cells, immortalized with the hybrid adenovirus/simian virus 40. H1198 and H1170 are both derived from BEAS cells; H1198 is pre-malignant, because 1) it is sensitive to serum in that it was growth inhibited and terminally differentiates into squamous cells similar to normal HBE and BEAS-2B cells; 2) in vitro invasiveness was detected after exposure of BEAS cells to either phorbol myristate acetate or cigarette smoke condensate (CSC). H1170 is defined as a malignant cell line because it exhibits several features that are typical of invasive adenocarcinomas, including increased expression of epidermal growth factor receptor (EGFR) and transforming growth factor-α (TGFα)[13]. We also investigated other non-small cell lung cancer (NSCLC) cell lines: H460, H1299, H596, H157, H552, H1944, H1792. The details of each cancer cell lines are given in Table 1.
Full table
We found that the expression levels of phospho-PRAS40 were higher in pre-maligment and maligmant cells than in normal cell lines (eg MCF10A); Later phase cancer cells, such as H1198/H1170, showed stronger PRAS40 activity than pre-cancer cell lines (eg BEAS) except H460, H1299, H1944, in which there were no p53 mutations. We also noticed that there was no difference in the total expression of PRAS40 and Akt, whereas the total expression of Raf was different in different types of cancer cell lines, for example, there was no expression in BEAS and H1944, and weak expression in H460 and H157 (Figure 4).
Discussion
The PI3K-Akt pathway has been implicated in the deve-lopment of multiple human cancers[14,15]. PI3K has an active role in oncogenic transformation. Akt, an important and probably essential downstream component of PI3K-mediated oncogenic signaling[16], provides a critical cell survival signal for tumor progression by phosphorylating a number of proteins involved in cell cycle regulation and proapoptotic factors[17]. Because only a subset of the cellular processes regulated by the PI3K-Akt pathway, are involved in tumorigenesis, the choice of drug targets must take into account the adverse effects resulting from the inhibition of other PI3K-Akt-dependent cellular processes. For example, the effects of insulin on metabolism are mediated through the PI3K-Akt pathway, so inhibitors of PI3K or Akt are therefore likely to perturb glucose homeostasis. It would be desirable, therefore, to target components of branches further downstream in the PI3K-Akt pathway[18].
Our data provided evidence that the proline-rich Akt-substrate PRAS40 showed higher expression levels in cancer cells (eg A549 and HeLa) than in normal cells (eg HEK293). In our breast cancer model (MCF10A/MCF7) and lung cancer model (BEAS/H1198/H1170) we also found the same result: PRAS40 was constitutively active in pre-malignant and malignant cancer cells (H1198/H1170 and MCF7), but only weakly expressed in normal cells (MCF10A and BEAS). In the NSCLC cell line, we found some interesting results: a higher expression level of phospho-PRAS40 was found in lung cancer cells with p53 mutations (eg H596, H157, H522 and H1792) than in other lung cancer cells. PRAS40 activity in lung cancer cells with only Ras mutations was almost the same as in the MCF10A normal cells (eg H460, H1299 and H1944). The strongest activity of PRAS40 was in H157, which had both the p53 mutation and the Ras mutation. The p53 mutation could be related to PRAS40 activity, but further studies are needed to clarify this relationship. These results suggested that PRAS40 activation is an early event during breast and lung carcinogenesis, PRAS40 could be used as a early detection marker in carcinogenesis.
We tested the effect of key kinase inhibitors on PRAS40 activity: PI3K inhibitors Worthmannin or Ly294002, MEK inhibitor Uo126 and mTOR inhibitor rapamycin. Only the PI3K inhibitors inhibited or decreased PRAS40 activity, therefore the PI3K-Akt survival pathway is the main pathway that PRAS40 is involved in; The Raf-MEK pathway probably doesn’t contribute to PRAS40 activity. Also, PRAS40 is the substrate of Akt, but it can be activated by an Akt- independent mechanism, for example, serum withdrawal decreases Akt activity, but has no effect on PRAS40.
14-3-3 is also a PRAS40 binding protein. A striking feature of the 14-3-3 proteins is their ability to bind a multitude of functionally diverse signaling proteins. This plethora of interacting proteins allows 14-3-3 to play important roles in a wide range of vital regulatory processes, such as mitogenic signal transduction, apoptotic cell death and cell cycle control[19]. Our data show that the expression level of 14-3-3, like that of PRAS40, is higher in the HeLa cell line than in HEK293, but exactly how 14-3-3 helps PRAS40 in the PI3K-Akt pathway is unknown and warrants further investigation.
To our knowledge, our results provide the first evidence that PRAS40 is constitutively active in pre-malignant and malignant breast and lung cancer cell lines, and that PRAS40 is mainly involved in the PI3K-Akt survival pathway. However, PRAS40 can also be activated by Akt- independent mechanisms. We suggest that PRAS40 could be chosen as an early detection marker in carcinogenesis, but it is also a protein that can be targeted by anti-tumor drugs, that is a “druggable” protein.
Acknowledgements
We thank Prof Hai-an FU for the generous gift of plasmids. We also thank the members of Prof Fu’s laboratory at Emory University for helpful discussions.
References
- Khuri FR, Herbst RS, Fossella FV. Emerging therapies in non-small-cell lung cancer. Ann Oncol 2001;12:739-44.
- Mc Williams A, Lam S. New approaches to lung cancer prevention. Curr Oncol Rep 2002;4:489-94.
- Goodman GE. Lung cancer 1: prevention of lung cancer. Thorax 2002;57:994-9.
- Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science 1997;278:1073-7.
- Vivanco I, Sawyers Cl. The phosphatidylinositol 3-kinase Akt pathway in human cancer. Nat Rev Cancer 2002;2:489-501.
- Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655-7.
- Manning BD, Cantley LC. United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapmycin (mTOR) signaling. Biochem Soc Trans 2003;31:573-8.
- Kovacina KS, Park GY, Bea SK, Gruzzetta AW, Schaefer E, Birnbaum MJ. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 2003;12:10189-94.
- Saito A, Narasimhan P. Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after Stroke: relationships with nerve growth factor. J Neurosci 2004;24:1584-93.
- Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1 induced cell death by 14-3-3 protein. Proc Natl Acad Sci USA 1999;96:8511-5.
- Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci USA 2001;14:7783-8.
- Goldman EH, Chen J, Fu H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at Ser967 and 14-3-3 dissociation. J Biol Chem 2004;279:10442-9.
- Chun KH, Kosmeder JW, Sun S, Pezzuto JM, Lotan R, Hong WK. Effect of deguelin on the phosphatidylinositol 3-Kinase/Akt pathway and apoptosis in premalignant human bronchial epithelial cells. J Nat Cancer Inst 2003;4:292-302.
- Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI3K-Kinase. Science 1997;276:1848-50.
- Klippel A, Escobedo MA, Wachowicz MS, Apell G, Brown TW, Giedlin MA, et al. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol 1998;18:5699-711.
- Toker A, Cantley L. Signaling through the lipid products of phosphoinositide-3-OH kinase. Nature 1997;387:673-6.
- Kobayashi M, Nagata S, Iwasaki T. Dedifferentiation of adenocarcinomas by activation of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 1999;96:4874-9.
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-akt pathway in human cancer: Rationale and promise. Cancer Cell 2003;4:257-62.
- Fu H, Subramanian HRS, Masters SC. 14-3-3 Proteins: structure, function, and regulation. Ann Rev Pharmacol Toxicol 2000;40:619-49.