
Role of arachidonic acid in hyposmotic membrane stretch-induced increase in calcium-activated potassium currents in gastric myocytes1
Introduction
Mechanical stretch is an important physiological stimulus in gut smooth muscles. It is well known that mechanical stretch induces myogenous contraction of gut smooth muscle, but the mechanism underlying this ionic channel process remains unknown. Mechanical stretch regulates the activities of ionic channels, which exist widely in the membranes of various cells and activate many signal transduction pathways. A hypothesis was proposed that membrane stretch induces alterations in the lipid bilayer, which transmits membrane tension to channel proteins or generates lipid-soluble second messengers, such as arachidonic acid (AA) and other endogenous fatty acids, by membrane-bound phospholipases[1,2]. Unsaturated fatty acids are major components of membrane lipids and they are mainly released by phospholipase A2 (PLA2) activation. AA in the cell membrane is esterified in phospholipids and can be released by PLA2 in response to various extracellular stimuli[3,4]. AA and other unsaturated fatty acids modulate the activities of various ion channels and enzymes through direct or indirect pathways. For example: AA potentiates hKir2.3 in part by decreasing inward rectification of the channel[5]; AA induces membrane depolarization by inhibiting KATP currents in murine colonic smooth muscle cells[6]; and AA increases choline acetyltransferase activity in spinal cord neurons, and this effect is mediated by protein kinase C (PKC)[7]. In addition, it was shown that AA induces endothelium-dependent hyperpolarization and relaxation of rabbit aorta through activation of apamin-sensitive K+ currents[8]. Abundant evidence has revealed that AA is an important mediator in hyposmotic stress. It was observed that swelling induces AA release via the 85 kDa cell phospholipase A2 (cPLA2) in human neuroblastoma cells[9], and that cell swelling activates PLA2 in ehrlich ascites tumor cells[10]. Tinel et al[11] also reported that AA acts as a second messenger for hypotonic-induced calcium transients in rat inner medullary collecting duct (IMCD) cells.
The calcium-activated potassium current (IK(Ca)) has been considered to play an important role in excitability and functional regulation in excitable cells. In our previous study, AA and other unsaturated fatty acids directly inhibited calcium currents[12] and muscarinic currents[13]. For both AA and hyposmotic membrane stretch-activated IK(Ca)[14,15], activation by hyposmotic membrane stretch is associated with calcium-induced calcium release (CICR)[16], which is triggered by extracellular calcium influx through the stretch-activated channels (SAC) in gastric antral circular myocytes of the guinea pig. However, the roles of AA and other unsaturated fatty acids in the process of IK(Ca) activation by membrane stretch in gastric myocytes remains unclear. In the present study, we therefore investigated the effects of AA and its metabolites on hyposmotic membrane stretch-induced increases in IK(Ca) in gastric antral circular myocytes of guinea pig.
Materials and methods
Preparation of cells EWG/B guinea pigs (obtained from the Experimental Animal Department of Norman Bethune University, Changchun, China; Certificate N
Electrophysiological recording Isolated cells were transferred to a small chamber (0.1 mL) on the stage of an inverted microscope (IX-70 Olympus, Tokyo, Japan) for 10–15 min to settle down. The cells were superfused continuously with isosmotic PSS by gravity (0.9–1.0 mL/min). An 8-channel perfusion system (L/M-sps-8, List Electronics, Darmstadt, Germany) was used to change solution. Experiments were carried out at 20–25 ºC and the whole-cell configuration of the patch-clamp technique was used. Patch-clamp pipettes were manufactured from borosilicate glass capillaries (GC 150T-7.5, Clark Electromedical Instruments, Kent, UK) using a 2-stage puller (PP-83, Narishige, Japan). The resistance of the patch pipette was 3–5 MΩ when filled with pipette solution. Liquid junction potentials were canceled before seal formation. Whole-cell currents were recorded with an EPC-10 patch-clamp amplifier (HEKA Instrument, Darmstadt, Germany) and command pulses were applied by using the Pentium IV-grade computer and pCLAMP software (Version 6.02; Axon Instruments, USA).
Drugs and solutions All drugs were purchased from Sigma (Sigma-Aldrich Corp St Louis, MO, USA). Tyrode’s solution contained 147 mmol/L NaCl, 4 mmol/L KCl, 1.05 mmol/L MgCl2·6H2O, 2 mmol/L CaCl2·2H2O, 0.42 mmol/L NaH2PO4, 1.81 mmol/L Na2HPO4·2H2O and 5.5 mmol/L glucose, and the pH was adjusted to 7.35 with NaOH. Ca2+-free solution contained 134.8 mmol/L NaCl, 4.5 mmol/L KCl, 10 mmol/L HEPES, 1 mmol/L MgCl2 and 5 mmol/L glucose, and the pH was adjusted to 7.4 with TRIZMA BASE (Tris). The isosmotic solution (290 Osmmol/kg) contained 80 mmol/L NaCl, 4.5 mmol/L KCl, 1 mmol/L MgCl2·6H2O, 2 mmol/L CaCl2·2H2O, 5 mmol/L glucose, 10 mmol/L HEPES and 110 mmol/L sucrose, and the pH was adjusted to 7.4 with Tris. Hypoosmotic solution (200 Osmmol/kg) contained 30 mmol/L sucrose, with the other ingredients at the same concentrations as in the isosmotic solution. Modified K-B solution contained 50 mmol/L L-glutamate, 50 mmol/L KCl, 20 mmol/L taurine, 20 mmol/L KH2PO4, 3 mmol/L MgCl2·6H2O, 10 mmol/L glucose, 10 mmol/L HEPES ,and 0.5 mmol/L egtazic acid, and the pH was adjusted to 7.4 with KOH. The pipette solution contained 110 mmol/L K-aspartic acid, 5 mmol/L Mg-ATP, 1 mmol/L MgCl2·6H2O, 20 mmol/L KCl, 0.1 mmol/L or 10 mmol/L egtazic acid, 2.5 mmol/L di-tris-creatine phosphate and 2.5 mmol/L disodium-creatine phosphate, and the pH was adjusted to 7.3 with KOH. AA, nordihydroguaiaretic acid (NDGA) and dimethyleicosadienoic acid (DEDA) were all prepared as aqueous stock solutions (1 mmol/L).
Data analysis Data were expressed as mean±SD. Statistical significance was evaluated using the Student’s t-test. Differences were considered to be statistically significant when P<0.05.
Results
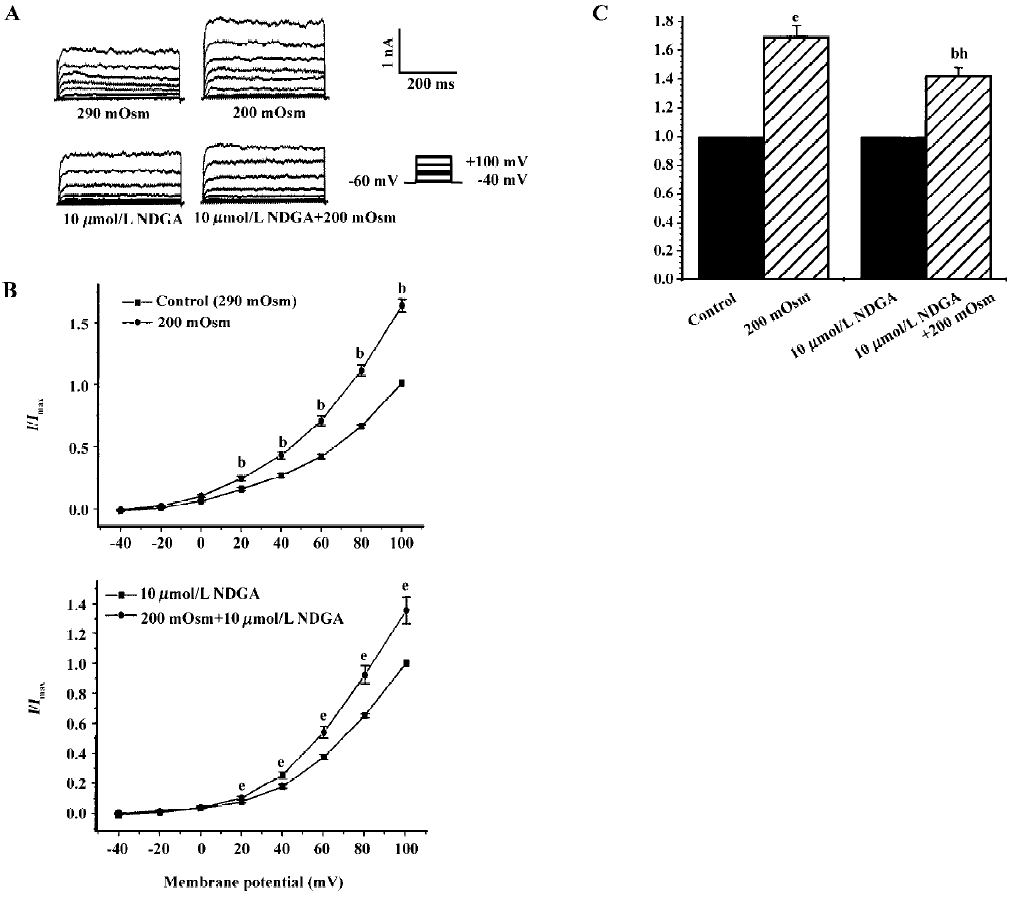
Effects of hyposmotic membrane stretch and AA on IK(Ca) and STOC Under whole-cell configuration, membrane potential was clamped at -60 mV, and IK(Ca) was elicited by a single-step command pulse from -60 mV to +60 mV for 400 ms at 15 s intervals. IK(Ca) started increasing at 139.3 s±11.3 s after cells were exposed to hyposmotic solution (200 mOsm), and at 165.0 s±25.1 s after cells were exposed to 10 µmol/L AA (Figure 1A,1B). There was no significant difference between the 2 latent periods. Hyposmotic membrane stretch and 10 µmol/L AA increased markedly in peak current of IK(Ca) to 168.3%±16.1% and 158.5%±20.5%, respectively (n=6, Figure 1B.).
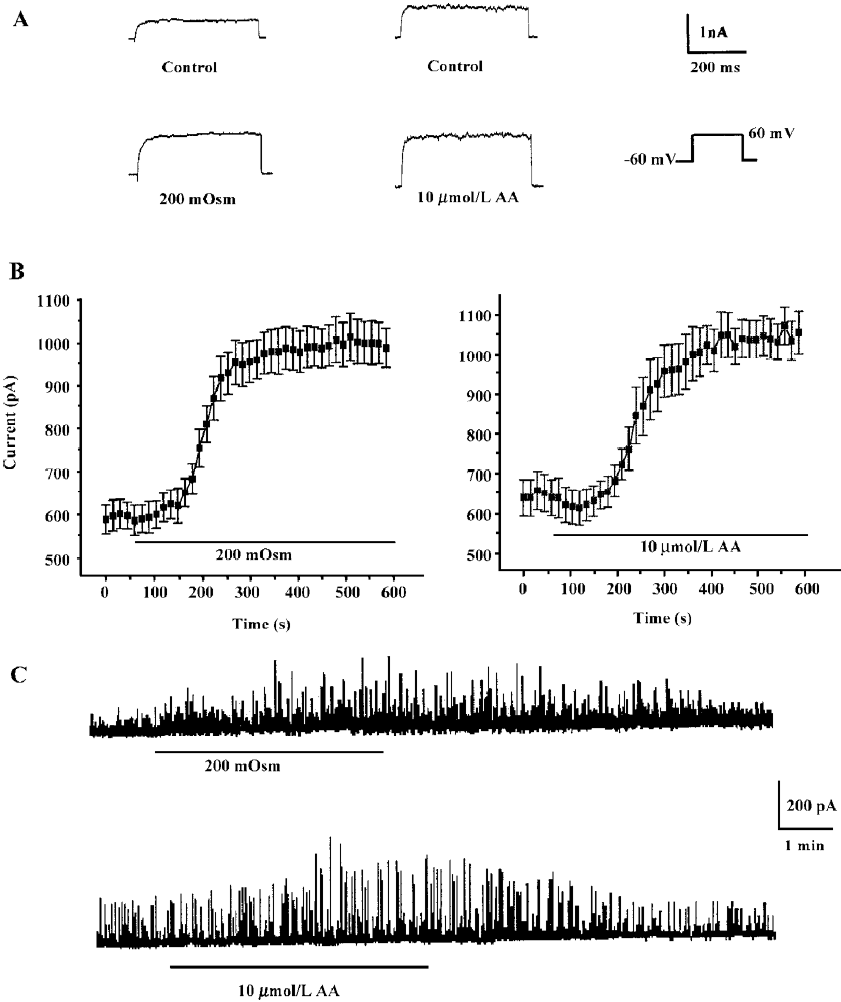
The calcium-activated potassium current is activated by intracellular Ca2+ and can be monitored by spontaneous transient outward currents (STOC)[17]. We therefore observed STOC to investigate effects of hyposmotic membrane stretch and AA on IK(Ca). In whole cell configurations, the holding potential was clamped at -20 mV, and STOC were elicited and enhanced by hyposmotic membrane stretch and 10 µmol/L AA, respectively (Figure 1C).
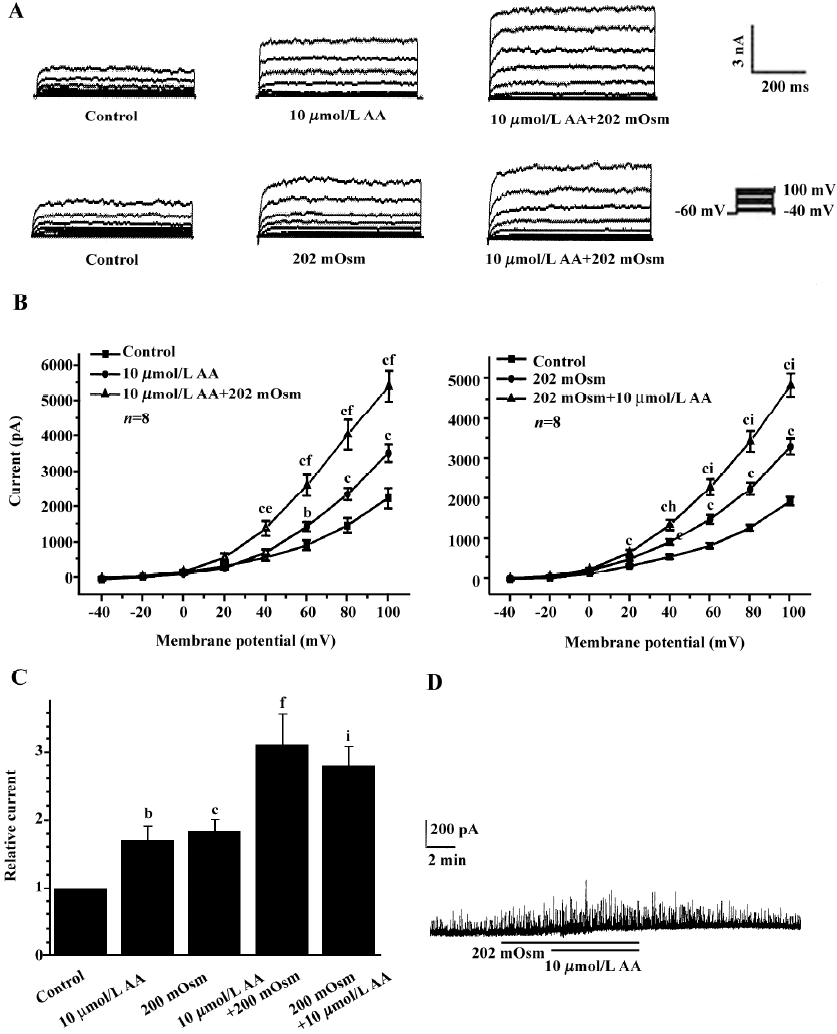
Effects of exogenous AA on hyposmotic membrane stretch-induced increase in IK(Ca) Under whole-cell configuration, membrane potential was clamped at -60 mV, and IK(Ca) was elicited by a step voltage command pulse from -40 mV to +100 mV for 400 ms with a 20-mV increment at 10-s intervals. Exogenous AA significantly increased IK(Ca) elicited by the command step pulse when membrane potential was depolarized from -40 mV to+100 mV, and hyposmotic membrane stretch potentiated the AA-induced increase in IK(Ca) when the membrane potential was depolarized from -40 mV to+100 mV (Figure 2A,2B). Hyposmotic membrane stretch also increased IK(Ca) elicited by the command step pulse when the membrane potential was depolarized from -40 mV to +100 mV, and AA potentiated hyposmotic membrane stretch-induced increase in IK(Ca) when membrane potential was depolarized from -40 mV to +100 mV (Figure 2A,2B). The peak current of IK(Ca) was increased to 184.2%±17.7% by hyposmotic membrane stretch and then the hyposmotic membrane stretch-induced increase in IK(Ca) was potentiated by 10 µmol/L AA, and the peak current increased to 281.3%±28.3% at +60 mV (n=8, Figure 2C). In another way, the peak current of IK(Ca) was increased to 171.8%±20.3 % by 10 µmol/L AA, and the AA-induced increase in IK(Ca) was potentiated by hyposmotic membrane stretch, with the peak current increasing to 311.5%±44.4% at +60 mV (n=8; Figure 2C). However, there was no significant difference between the potentiated effects of AA on the hyposmotic membrane stretch-induced increase in IK(Ca) and hyposmotic membrane stretch on the AA-induced increase in IK(Ca) (P>0.05, Figure 2C). As with the protocol above, the effects of hyposmotic membrane stretch and AA on STOC were investigated. Hyposmotic membrane stretch markedly increased STOC, and 10 µmol/L AA potentiated this effect (n=2, Figure 2D).
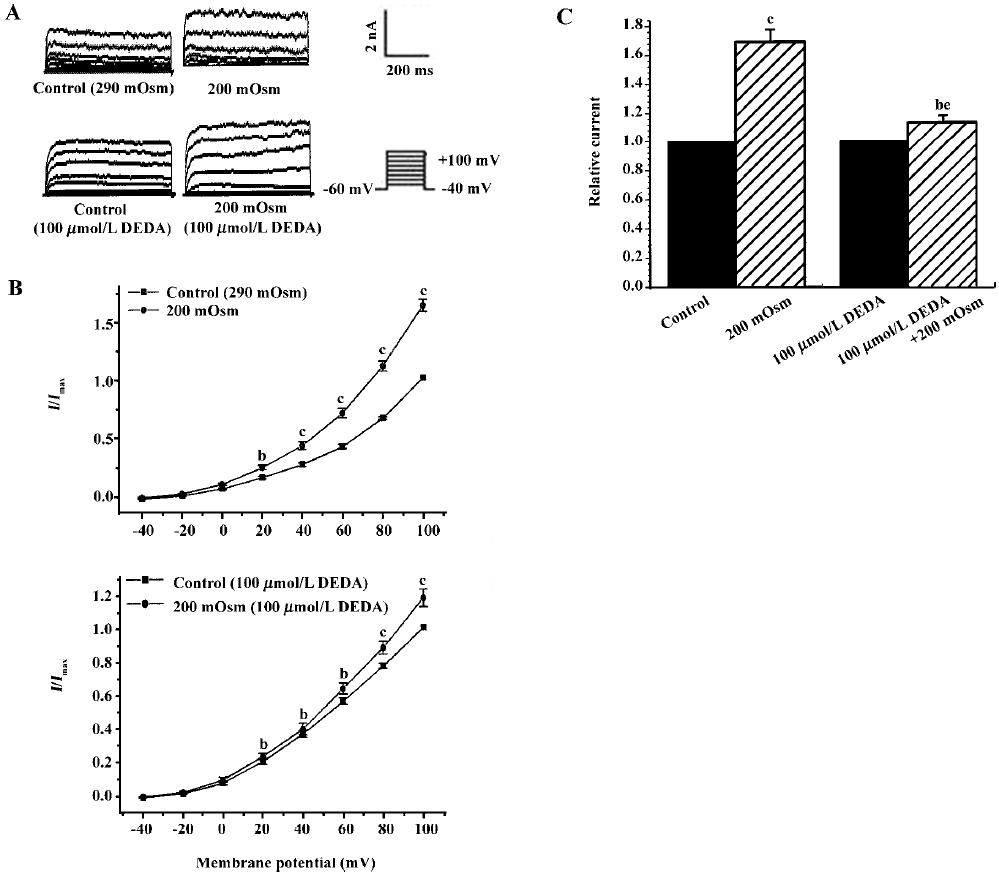
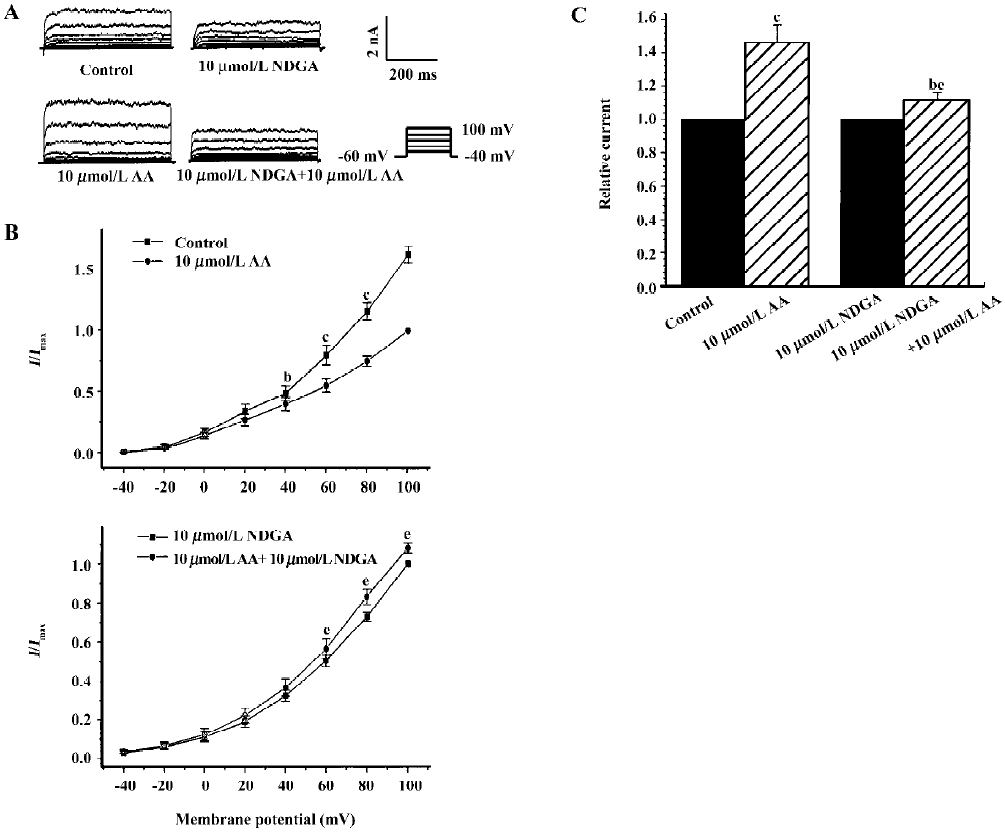
Effects of endogenous AA and its metabolites on hypos-motic membrane stretch-induced increase in IK(Ca) Intracellular free AA are metabolized by 3 enzymes: cycooxygenase, lipoxygenase and epoxygenase. To determine the effects of endogenous AA and its metabolites on the hyposmotic membrane stretch-induced increase in IK(Ca), DEDA, a non-selective inhibitor of PLA2, and NDGA, a lipoxygenase inhibitor, were used to inhibit the hydrolyzation of AA from membranes and to decrease the production of AA metabolites. DEDA (100 µmol/L in pipette) significantly suppressed the hyposmotic membrane stretch-induced increase in IK(Ca) (Figure 3A,3B), and the increased percentage was reduced from 164.3%±9.8 % of the control to 113.4%±3.6% at +60 mV (n=15, Figure 3C). When cells were pretreated for 15 min with 10 µmol/L NDGA, AA (Figure 4A,4B) and hyposmotic stretch-induced increases in IK(Ca) could be significantly suppressed (Figure 5A,5B). The AA-induced increase in IK(Ca) was decreased from 145%±10% to 110%±4% by NDGA (n=8, Figure 4C). NDGA also suppressed the hyposmotic membrane stretch-induced increase in IK(Ca) (Figure 5A,5B) and the increased percentage was reduced from 170%±10% to 142%±3% at +60 mV (Figure 5C). These results showed that endogenous AA and its metabolites were involved in the hyposmotic membrane stretch-induced increase in IK(Ca).
Effect of calcium mobilization on AA-induced increase in IK(Ca) It is well known that IK(Ca) is activated by intracellular free calcium, while extracellular calcium is necessary for efficiently controlling calcium homeostasis. To determine whether the AA-induced increase in IK(Ca) was mediated by calcium influx, the effect of AA on IK(Ca) was observed following the removal of extracellular calcium and the addition of 10 µmol/L EGTA in bath solution. The AA-induced increase in IK(Ca) was completely blocked by the removal of extracellular calcium, and the changes in the percentage of IK(Ca) were 146.30%±10.4% and 95.64%±11.7% in the presence or absence of extracellular calcium, respectively (n=6, Figure 6A). Our previous study indicated that hyposmotic membrane stretch activated L-type calcium currents[12] and calcium-activated potassium currents via extracellular calcium influx through SAC in gastric myocytes of guinea pig[15,16]. We therefore examined the relationship between the AA-induced increase in IK(Ca) and L-type calcium channels or SAC. However, 5 mmol/L nicardipine, an L-type calcium channel blocker, did not block the AA-induced increase in IK(Ca), but gadolinium (Gd3+), a blocker of SAC, completely suppressed the AA-induced increase in IK(Ca). The changes in the percentage of IK(Ca) were 146.3%±10.4%, 151.1%±14.4% and 102.5%±2.2% in the control, nicardipine and Gd3+ groups, respectively (n=6, Figure 6A).
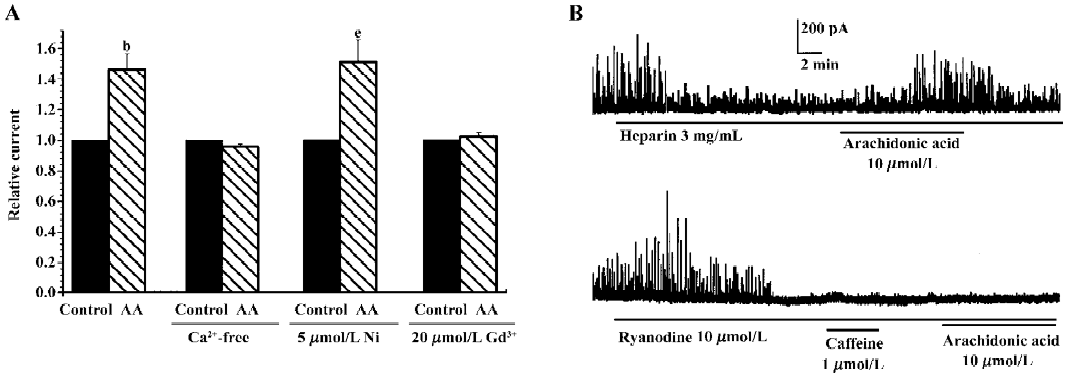
Intracellular free calcium has 2 sources: extracellular calcium influx and intracellular calcium release from calcium stores. Intracellular calcium is released through 2 pathways, one is CICR and the other is inositol-triphosphate-induced calcium release (IICR). We therefore investigated the role of intracellular calcium release in the AA-induced increase in IK(Ca). Heparin (3 mg/mL), a potent inhibitor of IICR, could inhibit STOC significantly, but did not block the AA-induced increase in STOC (Figure 6B). Ryanodine (10 µmol/L), a specific CICR inhibitor, binds to CICR channels and locks them in a subconductance state, thereby functionally depleting calcium stores[18]. In the present study, ryanodine increased STOC, and STOC were then almost abolished by ryanodine after approximately 8 min with caffeine, a CICR activator; AA could not then enhance them again (Figure 6B). These results indicated that CICR, but not IICR, was involved in the AA-induced increase in IK(Ca).
Discussion
Our previous study demonstrated that IK(Ca) was activated by hyposmotic membrane stretch and Ca2+ signaling played an important role in the process in gastric antral circular myocytes of guinea pig[15,16]. Under hyposmotic conditions, extracellular calcium influx through SAC triggered CICR, and intracellular free calcium then activated IK(Ca). However, it remains obscure how the membrane stretch is turned into the signal for Ca2+ entry from the extracellular space. We therefore investigated whether AA is involved in the hyposmotic membrane stretch-induced increase in IK(Ca).
In the present study, both hyposmotic membrane stretch and AA significantly increased IK(Ca) with a similar latent period. Moreover, exogenous AA potentiated the hypos-motic membrane stretch-induced increase in IK(Ca) (Figure 1). The results indicated that there may be a similar mechanism for the IK(Ca) activated by hyposmotic membrane stretch and AA. Activation of various signaling pathways may induce an increase in the production of AA, for example, phospholipase C, phospholipase D and PLA2. In mammalian tissues AA is mainly liberated directly from phospholipids by PLA2, which is a ubiquitous enzyme[4]. We have observed that when cells are exposed to DEDA, a non-selective inhibitor of PLA2, the hyposmotic membrane stretch-induced increase in IK(Ca) is significantly blocked by DEDA (Figure 3). The results suggest that hyposmotic membrane stretch activates PLA2, which hydrolyzes membrane phospholipids to produce AA, and AA as a second messenger mediates the hyposmotic membrane stretch-induced increase in IK(Ca) in gastric myocytes of guinea pig. Many experiments also support our results. It was observed that hyposmotic cell swelling induced AA release from cell membranes in human neuroblastoma cells[9] and Ehrilich ascites tumor cells[10]. In rat inner medullary collecting duct cells, AA acted as a second messenger in hypotonicity-induced calcium transients[11]. AA was also a second messenger in cultured rabbit principal cells[19] and ciliary epithelial cells under hyposmotic conditions[20]. Meanwhile, many reports have described that AA is able to affect cell functions, via its metabolites, under hyposmotic conditions in several cell systems. Leucotrienes, for example, appeared to mediate the inositol efflux in glial cells[21], and to activate chloride and potassium conductances as well as taurine transport in Ehrlich ascites cells[22]. In the present study, we also examined the possibility that AA metabolites could be involved in activating potassium currents under hyposmotic conditions by using NDGA, a lipoxygenase inhibitor. NDGA significantly inhibited AA and the hyposmotic membrane stretch-induced increase in IK(Ca) (Figure 4). These results suggest that AA metabolites generated by lipoxygenase mediate the hyposmotic membrane stretch-induced increase in IK(Ca) in gastric myocytes.
In various cell types, AA was found to induce Ca2+ flux and to mobilize intracellular calcium to trigger different Ca2+-dependent physiological functions in cells. For example, AA or its metabolites mobilized Ca2+ from intracellular stores, and intracellular Ca2+ then activated ion transport[19]. In several cell types, for example, in rat IMCD cells[11], human embryonic kidney (HEK293) cells[23] and Dictyostelium discoideum[24], AA released Ca2+ from the stores to trigger extracellular Ca2+ entry, and Ca2+ released from calcium stores was a prerequisite for extracellular Ca2+ entry. However, Murthy et al[20] found that AA induces Ca2+ influx, which triggers CICR in longitudinal smooth muscle of the intestine. Our previous study also demonstrated that hyposmotic membrane stretch activates IK(Ca) via CICR in gastric myocytes[15]. In the present study, the roles of AA and its metabolites in the relationships among hyposmotic membrane stretch-induced increase in IK(Ca), extracellular Ca2+ and intracellular calcium mobilization were investigated. Under extracellular calcium-free conditions, IK(Ca) was not increased by AA or hyposmotic membrane stretch (Figure 6). It was elucidated that extracellular calcium is necessary for AA and the hyposmotic membrane stretch-induced increase in IK(Ca), and some ionic channels participate in extracellular calcium influx. McCarty and O’Neil indicated that there are 2 alternative kinds of channel activated by hyposmotic swelling: voltage-activated Ca2+ channels and stretch-activated channels[25]. We observed previously that hyposmotic membrane stretch increases L-type current in gastric myocytes of guinea pig[12], and Yamamoto and Suzuki[26] also observed that there are 2 kinds of SAC in gastric myocytes of guinea pig. In the present study we examined whether these 2 channels are associated with extracellular calcium influx. Nicardipine, an L-type calcium channel blocker, could not block the AA-induced increase in IK(Ca). However, it was completely blocked by Gd3+, which blocks not only SAC but also store-operated Ca2+ channels (Figure 6). A similar effect of Gd3+ in blocking AA-induced Ca2+ entry has also been observed in other cell types, such as IMCD[11] and HEK293 cells[23].
Intracellular Ca2+ release from Ca2+ stores is the primary source of the increase in intracellular calcium. It was found that the entry of extracellular calcium via activating stretch-sensitive channels is amplified by calcium release from internal stores in toad gastric myocytes[26]. Sutko and Airey[27] suggested that ryanodine-sensitive calcium stores were positioned near the surface membrane in some smooth muscle cells, Ca2+ release from which was found to influence the activity of IK(Ca). Our previous study also indicated that hyposmotic membrane stretch activates IK(Ca)[15,16] and carbachol currents[13], and the activations are associated with CICR, which is triggered by extracellular calcium influx[15,28]. In the present study, heparin, a potent inhibitor of inositol triphosphate receptor, did not block the AA-induced increase in IK(Ca); however, ryanodine, a CICR agonist, completely blocked the AA-induced increase in IK(Ca) (Figure 6B). The results suggest that AA mobilizes intracellular calcium via triggering CICR and activates IK(Ca) in gastric antral circular myocytes of guinea pig. The potassium efflux through IK(Ca) hyperpolarized the membrane potential of smooth muscle cells, thereby limiting depolarization-dependent calcium and promoting relaxation. CICR can thus participate in both the contraction and relaxation of smooth muscle cells. Therefore, AA may be involved in both the contraction via activating extracellular Ca2+ influx and relaxation via activating IK(Ca) in gastric antral circular myocytes of the guinea pig.
In summary, hyposmotic membrane stretch may act on cell membranes to activate PLA2 and then generate AA. AA may then act as a second messenger to mediate extracellular calcium entry and trigger CICR to activate IK(Ca) in gastric myocytes of the guinea pig. AA and its metabolites may play an important role in regulating many cell functions under the hyposmotic conditions in gastric antral circular myocytes of the guinea pig.
References
- Morris CE. Are stretch-sensitive channels in molluscan cells and elsewhere physiological mechanotransducers? Experientia 1992;48:852-8.
- Ordway RW, Petrou S, Kirber MT, Walsh JV Jr, Singer JJ. Stretch activation of a toad smooth muscle K+ channel may be mediated by fatty acid. J Physiol 1995;484:331-7.
- Dennis EA, Rhee SG, Billah MM, Hannun YA. Role of phospholipase in generating lipid second messengers in signal transduction. FASEB J 1991;5:2068-77.
- Graber R, Sumida C, Nunez EA. Fatty acids and cell signal transduction. J Lipid Mediat 1994;9:91-116.
- Liu Y, Liu D, Krafte DS. Decrease of inward rectification as a mechanism for arachidonic acid-induced potentiation of hKir2.3. Eur Biophys J 2002;31:497-503.
- Jun JY, Yeum CH, Park YW, Jang IY, Kong ID, Sim JH, et al. Effects of arachidonic acid on ATP-sensitive K+ current in murine colonic smooth muscle cells. Jpn J Pharmacol 2002;90:81-7.
- Chalimoniuk M, King-Pospisil K, Pedersen WA, Malecki A, Wylegala E, Mattson MP, et al. Arachidonic acid increases choline acetyltransferase activity in spinal cord neurons through a protein kinase C-mediated mechanism. J Neurochem 2004;90:629-36.
- Gauthier KM, Spitzbarth N, Edwards EM, Campbell WB. Apamin-sensitive K+ currents mediate arachidonic acid-induced relaxations of rabbit aorta. Hypertension 2004;43:413-9.
- Basavappa S, Pedersen SF, Jorgensen NK, Ellory JC, Hoffmann EK. Swelling-induced AA release via the 85-kDa cPLA2 in human neuroblastoma cells. J Neurophysiol 1998;79:1441-9.
- Thoroed SM, Lauritzen L, Lambert IH, Hansen HS, Hoffmann EK. Cell swelling activates phospholipase A2 in Ehrilich ascites tumor cells. J Membr Biol 1997;160:47-58.
- Tinel H, Wehner F, Kinne RKH. Arachidonic acid as a second messenger for hypotonicity-induced calcium transients in rat IMCD cells. Pflügers Arch 1997;433:245-53.
- Xu WX, Kim SJ, Kim SJ, So I, Kang TM, Rhee JC, et al. Effect of stretch on calcium channel currents recorded from the antral circular myocytes of guinea-pig stomach. Pfluegers Arch 1996;432:159-64.
- Cui YF, Li L, Yu YC, Jin ZY, Li ZL, Xu WX. Role of unsaturated fatty acids in the enhancement of muscarinic current by hyposmotic membrane stretch in guinea pig smooth muscle cells. Acta Physiol Sin 2003;55:96-100.
- Zheng HF, Li XL, Jin ZY, Sun JB, Li ZL, Xu WX. Effect of unsaturated fatty acid on calcium-activated potassium current in guinea pig gastric myocytes. World J Gastroenterol 2005;11:672-5.
- Piao L, Li Y, Li L, Xu WX. Increment of calcium-activated and delayed rectifier potassium current by hyposmotic swelling in gastric antral circular myocytes of guinea pig. Acta Pharmacol Sin 2001;22:566-72.
- Piao L, Li Y, Li L, Ng J, Li ZL, Xu WX. The involvement of calcium mobilization in the calcium-activated potassium currents activated by hyposmotic swelling in gastric antral circular myocytes of the guinea-pig. Jpn J Physiol 2001;51:223-30.
- Bolton TB, Lim SP. Properties of calcium stores and transient outward currents in single smooth muscle cells of rabbit. J Physiol (Lond) 1988;409:385-400.
- Sutko JL, Airey J. Ryanodine receptor Ca2+ release channels. Physiol Rev 1996;76:1027-71.
- Ling BN, Webster CL, Eaton DC. Eicosanoids modulate apical Ca2+-dependent K+ channels in cultured rabbit principal cells. Am J Physiol 1992;63:F116-26.
- Civan MM, Coca-Prados M, Peterson-Yantorno K. Pathways signaling the regulatory volume decrease of cultured nonpigmented ciliary epithelial cells. Invest Ophthalmol Vis Sci 1994;5:2876-86.
- Strange K, Morrison R, Shrode L, Putnam R. Mechanism and regulation of swelling-activated inositol efflux in brain glial cells. Am J Physiol 1993;265:C244-56.
- Hoffmann EK, Dunham PB. Membrane mechanisms and intracellular signalling in cell volume regulation. Int Rev Cytol 1995;161:173-262.
- Luo D, Broad LM, Bird GSJ, Putney WJ Jr. Signaling pathways underlying muscarinic receptor-induced [Ca2+]i oscillations in HEK293 cells. J Biol Chem 2001;276:5613-21.
- Schaloske R, Sonnemann J, Malchow D, Schlatterer C. Fatty acids induce release of Ca2+ from acidosomal stores and activate capacitative Ca2+ entry in Dictyostelium discoideum. Biochem J 1998;332:541-8.
- McCarty NA, O’Neil RG. Calcium signaling in cell volume regulation. Physiol Rev 1992;2:1037-61.
- Yamamoto Y, Suzuki H. Two types of stretch-activated channel activities in guinea-pig gastric smooth muscle cells. Jpn J Physiol 1996;6:337-45.
- Sutko JL, Airey J. Ryanodine receptor Ca2+ release channels. Physiol Rev 1996;6:1027-71.
- Yu YC, Guo HS, Piao L, Li L, Lee ZL, Xu WX. Intracellular calcium involved in muscarinic currents increased by hyposmotic membrane stretch in gastric myocytes of guinea-pig. Acta Pharmacol Sin 2002;23:961-1056.