
Efficient inhibition of human telomerase activity by antisense oligonucleotides sensitizes cancer cells to radiotherapy1
Introduction
Human telomerase, activated in approximately 90% of cancers, is mainly composed of human telomerase RNA (hTR), human telomerase reverse transcriptase (hTERT) and telomerase-associated protein (TP1). hTR contributes by binding the telomere end through Watson-Crick base-pairing and acts as a template for polymerization, while hTERT catalyzes the extension of telomeric repeats. The determination of intracellular factors that control telomerase activity appears to be an important issue for cancer therapy. These observations suggest that reactivation of telomerase is necessary for proliferation of cancer cells and that inhibition of telomerase activity may be a useful strategy to suppress cell growth for many types of cancer[1,2]. The single-stranded DNA binding protein plays an important role in DNA replication, repair, and recombination. DNA double-strand breaks (DSB) are probably the most severe threat to the maintenance of genomic integrity. They are produced by ionizing radiation and some chemotherapeutic drugs, but also arise endogenously during DNA replication, by topoisomerase failure and as an initiator of physiological recombination processes like V (D) J recombination and meiosis. If left unrepaired, DSB can result in permanent cell-cycle arrest, induction of apoptosis, or mitotic cell death caused by the loss of genomic material [3,4].
Ionizing radiation (IR) is an important therapeutic modality in clinical cancer management, as it plays a key role in the treatment of many tumors. In several instances, sensitivity to cell killing by IR has been related to the greatly reduced ability to repair DNA DSB[5]. Animal models have shown that telomere dysfunction impairs DNA repair and thus enhances sensitivity to IR-induced cell killing. Although the loss of telomerase activity per se has no discernable impact on the response to ionizing radiation, the emergence of telomere dysfunction in late-generation hTR-/- mice imparts a radiosensitivity syndrome associated with accelerated mortality. On the cellular level, the gastrointestinal crypt stem cells and primary thymocytes show increased rates of apoptosis, and mouse embryonic fibroblasts (MEFs) show diminished dose-dependent clonogenic survival. The radiosensitivity of telomere dysfunctional cells correlates with delayed DNA break repair kinetics, persistent chromosomal breaks and cytogenetic profiles characterized by complex chromosomal aberrations and massive fragmentation. Furthermore, in vitro studies with C3H mouse acute myeloid leukemia cell lines have revealed that long but dysfunctional telomeres also correlate with IR sensitivity[6,7].
Previous studies targeting telomerase by antisense oligonucleotides against hTR as well as by pharmacological and genetic approaches indicate that telomerase is a promising target for cancer therapy. Indeed, preclinical evaluations of telomerase-based therapy show promising effects[8–11]. For blocking telomerase activity, one strategy is the design of agents that bind to the telomerase active site. The exposed RNA template of hTR is an ideal target for ASODN that can bind to telomerase, block telomerase activity, cause telomere shortening, and decrease cell proliferation[1,24]. Examining the effects of ionizing radiation on cells with siRNAs specific for hTERT, a further study has indicated that cells lacking hTERT showed a significantly increased sensitivity to ionizing radiation that induce DNA DSB[13]. Previous investigations[9,25,26] have suggested that telomerase inhibitor producing shortened telomeres in cancer cells selectively increase the radiosensitivity in tumour cells and therefore improve tumour killing through inducing DNA DSB and single strand breaks (SSB). However, the effect of the ASODN specific for hTR on radio sensitivity hasn’t been studied. The aim of our study was to investigate whether hTR-specific ASODN could be an efficient radiosensitizer and growth inhibitor, and to determine whether hTR-specific ASODN technology could serve as a method to affect telomerase-based cancer therapy.
Materials and methods
Cell lines and culture conditions Human glioma cell lines, U-251 cells were obtained from the Wuhan University type culture collection. U-251 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), and incubated at 37 °C in the humidified atmosphere containing 5% CO2.
Antisense and nonspecific oligonucleotides transfection The sequence of the hTR ASODN was 5'-CAGTTAGGGTTAG- 3', complementary to bases 38–50 of hTR, which was referred to Eckart et al[12]. The sequences 5'-CTAACCCTAACTG-3' were used as nonspecific oligonucleotides (NSODN). The oligonucleotides used were end phosphorothioated and synthesized in TaKaRa Bio Inc (Shiga, Japan). To confirm the above gene sequences as unique (not consistent with other gene sequences), we conducted a BLAST (Basic Local Alignment Search Tool) search of the IMAGE EST (Express-ed Sequence Tags) library (www.ncbi.nlm.nih.gov/BLAST).
These oligonucleotides were transfected into U-251 cells with lipofectamine reagent (Gibco Inc, USA) according to the manufacturer’s instructions. The final concentration of oligonucleotides was adjusted to 3 μmol/ L and the final concentration of lipofectamine was 10 μmol/L. A defined number of cells (3×105–5×105) were seeded in 25 cm2 flasks, 80%–90% confluent at the time of transfection. The antisense or nonspecific oligonucleotides and lipofectamine reagent were mixed and preincubated for 15 min at room temperature in serum- and antibiotic-free RPMI-1640 respectively, then mingled for 20 min. The final volume of oligonucleotides mixture in every flask was 3 mL, containing 48 μg oligonucleotides and 120 μL lipofectamine reagent. U-251 cells were treated with the oligodeoxynucleotide mixture at 37 °C in a CO2 incubator until ready to assay (24 h post-transfection).
Ionizing radiation After transfection, the cells were irradiated at room temperature with a single dose of 0 or 2Gy and the dose rate was 2 Gy/min at room temperature using a 60-Cobalt source. As further controls, cells without transfection treated with the same irradiation regime were investigated. Cells were harvested at 1, 2 and 6 h after irradiation for further analysis of telomerase activity, telomere length and Comet Assay.
Telomerase activity assay The telomerase activity was determined using the PCR-TRAP ELISA kit (Roche, Indianapolis, IN, USA) according to the manufacturer’s description with some modifications. To determine the telomerase activity, the cells were harvested and counted. One million cells were taken in an Eppendorf tube and used per reaction. Cells were pelleted at 3 000 ×g for 10 min in a refrigerated centrifuge at 4 °C. Cells were washed three times with cold phosphate buffered saline and processed as described in the protocol supplied with the kit. For one million cells 100 μL pre-cooled lysis reagent (solution 1 of the kit) was added and incubated on ice for 30 min and centrifuged at 15 000×g for 20 min at 4 °C. Supernatant was collected, protein concentrations were determined, and various concentrations of cell extract were used for the telomerase assay using the Telomerase PCR ELISA procedure. Telomeric repeats were added to a biotin-labeled primer by telomerase, contained in the sample, followed by amplification of the elongation product by PCR. The product was aliquoted, denatured, bound to a streptavidin-coated 96-well plate, and hybridized to a DIG-labeled telomeric repeat-specific probe. An antibody to DIG, conjugated to peroxidase, was subsequently bound to DIG and visualized by virtue of the enzyme’s ability to metabolize tetramethyl benzidine (TMB) so as to produce a colored reaction product. The absorbance of the samples was measured at 450 nm using an ELISA microtiter plate reader within 30 min after addition of the stop reagent.
Telomere length analysis Approximately 0.5 to 1 million cells were used for the preparation of high-molecular-weight DNA by DNA extraction kit. Mean length of terminal restriction fragment (TRF) was measured using the TeloTAGGG telomere length assay kit (Roche Molecular Biochemical, Indianapolis, IN, USA). Two μg of purified DNA was digested with Hinf I/Rsa I mixture (10 U/ μg DNA) for 2 h, separated on 0.8% agarose gel electrophoresis in 1×Tris-acetate-EDTA(-ethylenediaminetetraacetic acid), and transferred onto a nylon membrane according to the protocol described by the supplier. After rinsing in 2 ×SSC and neutralizing solution, the filter was hybridized to digoxigenin-labeled probe specific for telomeric repeats for 3 h at 42 °C. The filter was washed twice in 2 ×SSC/0.1% sodium dodecyl sulfate (SDS) for 5 min each time at room temperature and then was placed in 0.2 ×SSC/0.1% SDS solution at 50 °C for 15 min with one repeat. To determine TRF length, the hybridized probe was incubated with a digoxigenin-specific antibody covalently coupled to alkaline phosphatase. Finally, the immobilized telomere probe was visualized by a highly sensitive chemiluminescent substrate for alkaline phosphatase, CDP-Star, and was used to detect TRF DNA on Hyperfilm. Overall mean TRF length was determined as described previously. In addition, mean TRF of the longer telomeres (longer than the overall mean TRF distribution) and mean TRF of the shorter telomeres (shorter than the overall mean TRF distribution) were calculated using MacBasV-2.5 software(Fuji, Stamford, Connecticut, USA).
Comet assay DNA damage was evaluated using the neutral single-cell gel electrophoresis or comet assay. Harvested cells (1×105) were mixed with 0.5% low melting agarose (Gibco) and layered onto agarose-coated slides. Slides were then submerged into lysis buffer (30 mmol/L Na2EDTA, 0.5% SDS; pH 8.3) overnight at 4 °C. After lysis, slides were incubated for 10 h in electrophoresis buffer. After electrophoresis (0.6 V/cm, 25 min), slides were neutralized with 0.4 mol/L Tris, pH 7.5, twice per 5 min, and placed into 100% ethanol and then air-dried. Slides were stained with 2 µg/mL ethidium bromide (Sigma). Finally, slides were observed by confocal microscopy. Average Comet Tail Moment was scored by using the Comet Imager software (IMI1.0) (Liangzheng software Co Ltd, Beijing, China). All measurements were done on duplicate slides.
Clonogenic assay Cell survival after ionizing radiation treatment was determined by clonogenic assay. Depending on the optimal plating efficiency for U-251 cell line, approximately 500 to 30 000 cells were seeded in 25 cm2 flasks. Cells, irradiated with1, 2, 3, 4, 6, 8, or 10 Gy of ionizing radiation, were incubated at 37 °C in 5% CO2 for 12 d followed by fixation with acetic acid: methanol (1:3), stained by crystal violet and counting of the surviving clones. Cell survival was measured by standard colony formation after radiation treatment. Colonies containing >50 cells were rated as deriving from viable, clonogenically capable cells.
Statistical analysis Statistical analysis of the results was carried using an SSPS 13.0 package. Differences between groups were evaluated by Student’s t-test. GraphPad execute curve fitting of survival fraction. A P-value of <0.05 was considered to be statistically significant.
Results
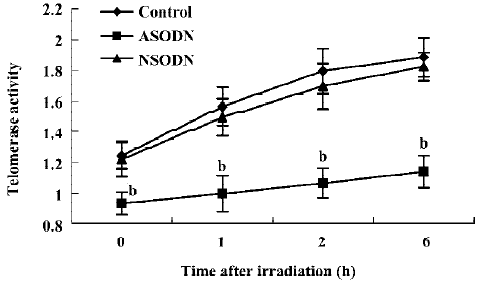
Telomerase activity inhibition by ASODN against hTR In this study, the ASODN targeting hTR was transfected into U-251 cells mediated by lipofectamine, and which resulted in a significantly decreased telomerase activity as measured by the TRAP assay. Cells treated with nonspecific oligonucleotides (NSODN) showed telomerase activity equivalent to that of the control group. Furthermore, telomerase activity of the three groups mentioned above was also measured at the 1st, 2nd, and 6th hour after IR treatment, which showed that telomerase activity of cells in ASODN groups decreased significantly compared to that in cells of other groups exposed to IR. It indicated that stable suppression of hTR by ASODN functionally inhibits telomerase activity in human cancer cells with or without IR exposure (Figure 1).
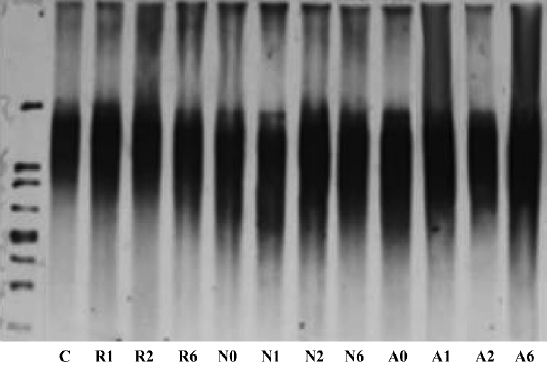
We then examined the effects of suppressing hTR on telomere structure (Figure 2). Analysis of terminal restriction fragments (TRF) by Southern blotting demonstrated that telomere lengths observed in ASODN groups (A0, A1, A2, A6) were 7–8 kb, similar to those of both the NSODN treated group (N0, N1, N2, N6) and control group (C). These results indicated that there was no effect of hTR ASODN on telomere structure over a short period of time.
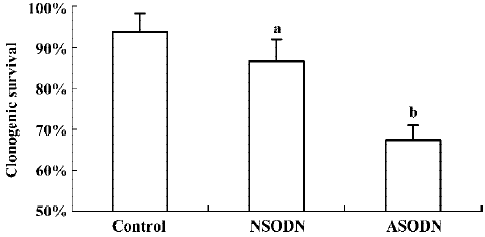
Cell growth inhibition by ASODN based suppression of hTR To determine if ASODN based suppression of hTR had any effects on U251 cells, clonogenic survival was analyzed without IR treatment (Figure 3). The clonogenic plating efficiency of ASODN group cells (67.20%) was obviously lower than that of control group cells (93.80%) as well as NSODN group cells (83.60%). Compared with control cells, a slight decline in clonogenic survival in NSODN group cells was also observed. Our observations indicated that treatment with ASODN had an effect on cell growth inhibition in U251 cells; meanwhile this effect was also detected on NSODN treated cells but more gently.
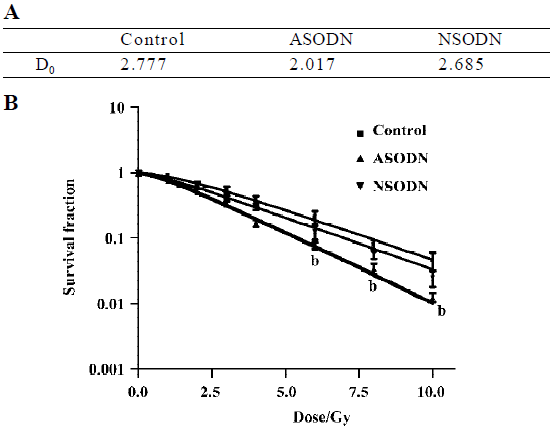
Increased sensitivity of hTR-inhibited cells to ionizing radiation The core of our study was to determine whether hTR-specific ASODN technology could serve as a method to affect telomerase-based cancer therapy. Telomerase-based therapies were expected to have the greatest use when combined with canonical cancer therapies, such as radiotherapy. We thus examined the clonogenic survival of all above-mentioned groups after exposure with 1, 2, 3, 4, 6, 8, 10 Gy doses of γ-rays (Figure 4). Based on the equation with single-target multi-hit model, the reduction of D0 value represented the increase of radiosensitivity. In our results, the D0 value of hTR-specific ASODN group was lower than that of control group and NSODN group (Figure 4A), which suggested that ASODN-based hTR suppression increased radiosensitivity. It showed that clonogenic survival of the ASODN groups was the lowest among three groups at various doses in Gy, when comparing ASODN treated groups with control group and NSODN treated group, respectively (Figure 4B).
We further examined the effects of ASODN-based suppression of hTR on radiosensitivity. To confirm the role of ASODN in DNA repair in U251 cells, we measured DSB proficiency in individual cells by comet assay (Figure 5A, 5B). DNA damage and the residual levels were assessed in the cells before, and at 1, 2 and 6 h after 2 Gy irradiation. The results of DNA tail in percent were shown in Figure 5A. DNA tail of control, ASODN and NSODN groups were at the same level before exposure (shown as 0 h), and that of ASODN groups were more than other groups after exposure at all time points. In spite of DNA tail increase of all groups after 2 Gy dose IR treatment, the curve of ASODN groups was obviously changed to higher levels indicating that the effect of hTR-specific ASODN was an increase in level of DSB after exposure to IR. These findings suggested that the effects of hTR-inhibited treatment with ASODN could improve radiosensitivity of U251 cells immediately after IR exposure. The results of both comet assay and clonogenic survival assay demonstrated a suppression of hTR expression by ASODN in U251 cells, leading to increased sensitivity to radiation.
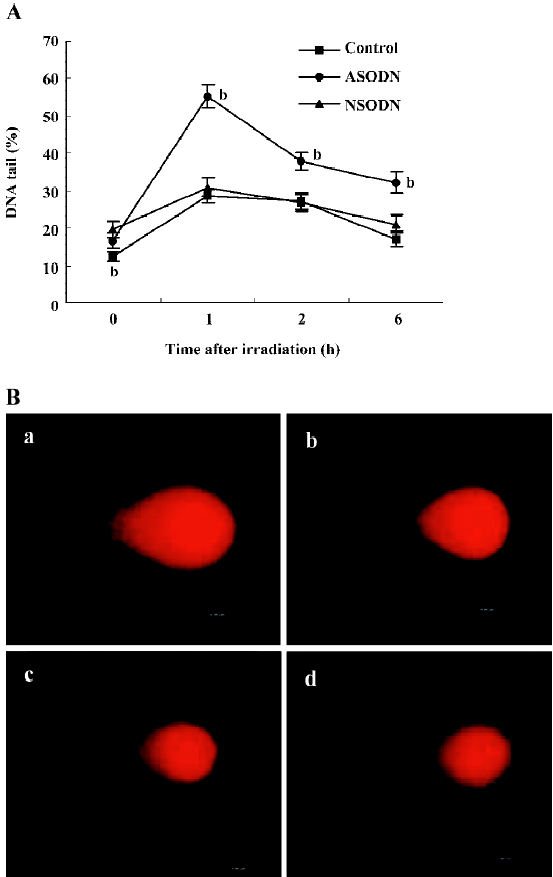
Discussion
The overall 5-year survival for neurogliocytoma has not changed significantly during the past 20 years. A promising target for new approaches to cancer therapy has emerged recently in the form of the enzyme telomerase[14]. The role of telomeres in radiosensitivity has recently surfaced. When telomerase is inactivated in mice by the targeted disruption of the hTR gene, the telomeres progressively shorten with each generation until a radiosensitivity syndrome is induced, leading to accelerated mortality on exposure to ionizing radiation[6,10,17]. Another study showed that fifth-generation telomerase-deficient mice, which have telomeres 40% shorter than wild-type control animals, are hypersensitive to ionizing radiation. The mice usually die of acute radiation poisoning in the gastrointestinal tract, lymphoid organs, and kidneys. The telomerase-deficient animals show greater chromosomal damage and higher numbers of apoptotic cells than the similarly irradiated control mice[15]. These findings indicate that components of telomerase are attractive targets for improved radiosensitivity.
Previous studies, targeting the reduction of telomerase activity by ASODN against hTR or hTERT, have shown promising effects[6,10,13,17]. Despite these findings, other studies have suggested a more direct interaction between hTERT and the DNA repair process in human cells. They indicate increased sensitivity of cells inhibiting hTERT to ionizing radiation that induces DNA strand breaks[13,18]. Using ASODN targeting at hTR, we find that U-251 cells exhibit significantly decreased telomerase activity. Although another study suggests that expression of antisense hTR results in a gradual reduction in telomere length[19], we have not detected alterations in overall telomere length over a short time period, in keeping with a late finding of antisense hTERT[18]. After treatment with ASODN, we have detected a significant reduction of clonogenic growth in ASODN transfected group than in the control groups. Similarly, another investigation has shown that prolonged treatment with 2-O-methoxyethyl RNA oligomers against hTR cause prostate cancer cells becoming nonviable after 60–80 d[17]. Further-more, at least one study has observed that suppression of hTERT expression alters the proliferative potential of human cancer cells, leading to decreased tumorigenic potential[13]. Our findings suggest that, even partial reduction in growth rates during treatments with hTR; ASODN in vitro may have therapeutic values for patients.
Importantly, suppression of hTR expression by ASODN has shown increased radiosensitivity in our experiments. After treatment with IR, we observed an obvious increase of DSB in U-251 cells transfected with hTR ASODN compared to other groups. After several hours of DNA repair, the same increase of DSB in the hTR ASODN group can be detected. Although another investigation has reported that the radiosensitivity of telomere dysfunctional cells correlates with delayed DNA break repair kinetics, persistent chromosomal breaks and cytogenetic profiles characterized by complex chromosomal aberrations and massive fragmentation[6], our study has shown a more direct effect of hTR ASODN on radiosensitivity. All these findings suggest that telomerase-inhibitor therapy should selectively increase the radiosensitivity of the tumour and therefore improve tumour killing.
Oligomers that target hTR are more potent inhibitors than those targeting hTERT mRNA. This finding is important because it is consistent with the belief that the telomerase RNA template is an exceptionally promising target for oligonucleotide-mediated inhibition. They explain that the template region is unusually accessible to hybridization because of the requirement for binding telomere ends. The localization of telomerase to the nucleolus may also play a role, possibly by promoting colocalization of oligonucleotides and targets. Conversely, the mRNA encoding hTERT may be an unusually difficult target because it has an unusually high percentage of cytosine and guanine residues (66%). This is especially true within the first 1000 bases, which are characterized by a C/G content of 75%[20]. Although telomerase activity requires coexpression of hTR and hTERT components, hTERT expression is correlated more closely with telomerase activity in cancer cells[21,22] and most studies have shown advantages in strategies targeted at hTR[16,23]. Our findings have shown efficiency of hTR ASODN, which is an encouraging perspective for cancer therapy.
Interestingly, although a previous finding indicated that telomere dysfunction correlates with radiosensitivity[6], we have observed an increase in radiosensitivity before telomere length shortens after ASODN transfection, which implies, a yet unknown potential mechanism through which hTR ASODN may increase radiosensitivity. Meanwhile, in our experiments, the decrease of telomerase activities after ASODN transfection can be detected. Future studies are necessary to clarify if the reduction of telomerase activity is the cause of increased radiosensitivity.
In summary, hTR ASODN, the inhibitor for telomerase activity, has produced a direct therapeutic value and increased radiosensitivity in cancer cells, therefore improving cancer killing. Compared with hTERT inhibitor, hTR ASODN showed that targeting hTR might be more potent with simple manipulation. Furthermore, this may allow the use of lower doses of ionizing radiation, thus achieving greater and selective tumour killing with less severe side effects in radiotherapy. Regardless of the mechanisms, our data support the objective of inducing radiation sensitivity through targeting hTR.
References
- Natarajan S, Chen Z, Wancewicz EV, Monia BP, Corey DR. Telomerase reverse transcriptase (hTERT) mRNA and telomerase RNA (hTR) as targets for downregulation of telomerase activity. Oligonucleotides 2004;14:263-73.
- Liu JP. Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J 1999;13:2091-104.
- Rothkamm K, Löbrich M. Misrepair of radiation-induced DNA double-strand breaks and its relevance for tumorigenesis and cancer treatment. Int J Oncol 2002;21:433-40.
- Purnapatre K, Varshney U. Cloning, over-expression and biochemical characterization of the single-stranded DNA binding protein from mycobacterium tuberculosis. Eur J Biochem 1999;264:591-8.
- Pandita TK, Roti JL. Role of telomerase in radiocurability. Onc Rep 2003;10:263-70.
- Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, et al. Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet 2000;26:85-8.
- Finnon P, Silver AR, Bouffler SD. Upregulation of telomerase activity by X-irradiation in mouse leukaemia cells is independent of Tert, Terc, Tnks and Myc transcription. Carcinogenesis 2000;21:573-8.
- Norton JC, Piatyszek MA, Wright WE, Shay JW, Corey DR. Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol 1996;14:615-9.
- Kondo Y, Koga S, Komata T, Kondo S. Treatment of prostate cancer in vitro and in vivo with 2-5A-anti-telomerase RNA component. Oncogene 2000;19:2205-11.
- Chen Z, Koenemen KS, Corey DR. Consequences of telomerase inhibition and combination treatments for the proliferation of cancer cells. Cancer Res 2003;63:5917-25.
- Corey DR. Telomerase inhibition, oligonucleotides, and clinical trials. Oncogene 2002;21:631-7.
- Eckart M, Christine L, Marianne S, Yong W, Lora D, Eugen U, et al. Potent inhibitory activity of chimeric oligonucleotides targeting two different sites of human telomerase. Oligonucleotides 2005;15:255-68.
- Nakamura M, Masutomi K, Kyo S, Hashimoto M, Maida Y, Kanaya T, et al. Efficient inhibition of human telomerase reverse transcriptase expression by RNA interference sensitizes cancer cells to ionizing radiation and chemotherapy. Hum Gene Ther 2005;16:859-68.
- McCaul JA, Gordon KE, Clark LJ, Parkinson EK. Telomerase inhibition and the future management of head-and-neck cancer. Lancet Oncol 2002;3:280-8.
- Goytisolo FA, Samper E, Martin-Caballero J, Finnon P, Herrera E, Flores JM, et al. Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J Exp Med 2000;192:1625-36.
- Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, Blackburn EH. A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci USA 2001;98:7982-7.
- Fu XH, Zhang JS, Zhang N, Zhang YD. Combination of telomerase antisense oligonucleotides simultaneously targeting hTR and hTERT produces synergism of inhibition of telomerase activity and growth in human colon cancer cell line. World J Gastroenterol 2005;11:785-90.
- Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, et al. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci USA 2005;102:8222-7.
- Mo Y, Gan Y, Song S, Johnston J, Xiao X, Wientjes MG, et al. Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res 2003;63:579-85.
- Natarajan S, Chen Z, Wancewicz EV, Monia BP, Corey DR. Telomerase reverse transcriptase (hTERT) mRNA and telomerase RNA (hTR) as targets for downregulation of telomerase activity. Oligonucleotides 2004;14:263-73.
- Kim SH, Kamainker P, Campisi J. Telomeres, aging and cancer: In search of a happy ending. Oncogene 2002;21:503-11.
- Poole JC, Andrews LG, Tollefsbol TO. Activity, function, and gene regulation of the catalytic subunit of telomerase (hTERT). Gene 2001;269:1-12.
- Bisoffi M, Chakerian AE, Fore ML, Bryant JE, Hernandez JP, Moyzis RK, et al. Inhibition of human telomerase by a retrovirus expressing telomeric antisense RNA. Eur J Cancer 1998;34:1242-9.
- Zhang P, Xu Q, Chen WT, Duan LQ, Zhang ZY, Zhou XJ. Synergistic down-regulation of telomerase by all-trans retinoic acid and antisense oligonucleotide in oral squamous cell carcinoma cell line (Tca8113). Oral Oncol 2005;41:909-15.
- Norton JC, Piatyszek MA, Wright WE, Shay JW, Corey DR. Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol 1996;14:615-9.
- Chen Z, Koeneman KS, Corey DR. Consequences of telomerase inhibition and combination treatments for the proliferation of cancer cells. Cancer Res 2003;63:5917-25.