
Effects of (-)stepholidine in animal models for schizophrenia1
Introduction
(-)Stepholidine (SPD) is one of the active ingredients of the Chinese herb Stephania intermedia and belongs to the tetrahydroprotoberberines (THPBs)[1], which consist of a four-ring main structural backbone with substituents at the two benzene rings (Figure 1). SPD is characterized by two –OH in the C2 and C10 position and two –OCH3 in the C3 and C9 position. Receptor binding studies have shown that SPD has a relative high affinity for the D1 (Ki of 13 nmol/L) and less affinity for the D2 (Ki of 85 nmol/L) receptors[2]. Electrophysiological studies have shown that SPD induces depolarization inactivation of dopaminergic cells in the ventral tegmental area (A10) but not in the substantia nigra (A9)[3]. This pattern is characteristic of some atypical antipsychotics, such as clozapine and thioridazine[4,5], although the newer antipsychotics such as risperidone and ziprasidone do not seem to selectively affect the A10 cell group[6]. Moreover, SPD also shows antipsychotic-like activity in several behavioral experiments. Thus it reverses apomorphine-induced stereotyped behaviour and induces a weak, short-lasting catalepsy[7]. In addition, SPD reverses amphetamine-induced or D2 selective agonist-induced rotations in 6-OHDA lesioned rats[8] and increases serum prolactin levels[9]. In an experiment with forskolin-induced stimulation of adenylate cyclase activity, SPD did not affect the formation of cAMP, but it reversed the dopamine-induced inhibition, which is mediated via D2 receptors[10]. SPD also reverses the apomorphine-induced inhibition of firing of the A9 dopaminergic neurons[11] and of A10[12]. All of these data point to a dopamine D2 antagonistic effect of SPD.
Interestingly, by itself SPD induces contralateral rotations and enhances the effects of apomorphine in 6-OHDA lesioned rats, suggesting an agonistic action[8]. In addition, SPD increases the production of cAMP by stimulating the Gs protein [13], and increases the phosphorylation of DARPP-32[14]. This strongly suggests that in addition to its D2 antagonistic properties, SPD is a dopamine D1 agonist, thus giving it a unique pharmacological profile. Further studies have shown that SPD has D1 agonistic effects on the firing activity in the medial prefrontal cortex[15] and D1 agonistic/D2 antagonistic effects in the nucleus accumbens[16]. Taking into account all of these data, it can be suggested that SPD represents a novel antipsychotic drug. Indeed preliminary clinical studies seem to underline this presumption[17].
The aim of the present study was to investigate whether SPD indeed shows antipsychotic-like actions in established animal models for schizophrenia. Therefore we compared the effects of SPD with those of haloperidol and clozapine in two different behavioural paradigms: (1) the paw test and (2) drug-induced disruption of prepulse inhibition. The paw test was developed approximately 20 years ago in an attempt to differentiate between classical and atypical antipsychotics[18]. In this test, a rat is placed on a platform with its fore- and hindlimbs extended through four holes. The dependent parameters are the retraction times for the fore- and the hindlimbs. Results from a large series of pharmacological studies show that all antipsychotics increase the hindlimb retraction time, and only the classical antipsychotics affect the forelimb retraction time[19–21], thus allowing a differentiation between classical and atypical antipsychotics.
The reversal of a drug-induced prepulse inhibition deficit is an extensively used model for identifying (novel) antipsychotic compounds. Although many different drugs can disrupt prepulse inhibition, especially the disruption induced by dopamine agonists or glutamate antagonists has been used in antipsychotic research[22]. With respect to the former, it has been shown that all antipsychotics reverse the prepulse inhibition deficit induced by dopamine agonists such as apomorphine[22,23]. The reversal of glutamate antagonist induced disruption by antipsychotics is less obvious. Thus, whereas most authors agree that classical antipsychotics are ineffective[24–28], some[29–31] but not all[24,26,32,33] have found that clozapine can reverse the effects of glutamate antagonists.
Material and methods
Rats and housing Male Wistar rats weighing between 230 and 300 g were used (Central Animal Laboratory, Nijmegen, the Netherlands). The rats were housed in groups of 2 or 3 rats in standard Macrolon cages until the day prior to the experiment, in temperature controlled rooms (23±1 oC). The rats were maintained on a standard 12 h light-dark cycle (lights on from 7:00 AM to 7:00 PM), and had free access to food and water, except during the experiment. Animals were only used once throughout the experiment.
Paw test The paw test was performed using a Perspex platform measuring 30 cm×30 cm, with a height of 20 cm. The top of the platform had two smaller holes of 4-cm diameter for the forelimbs, two larger holes of 5 cm diameter for the hindlimbs and a slit for the tail[18]. The paw test was performed 30 min after intraperitoneal administration of the solvent or the drug by taking the rat behind the forelimbs and carefully lowering the hindlimbs in the holes, followed by the forelimbs. The Forelimb Retraction Time (FRT) was defined as the time it took the rat to withdraw one forelimb. Likewise, the Hindlimb Retraction Time (HRT) was defined as the time it took the rat to withdraw one hindlimb. For both FRT and HRT the minimum was set to 1 s and the maximum to 30 s. The paw test was repeated at 40 and 50 min after the injection. As there were no statistically significant differences between the scores at 30, 40 and 50 min, the average FRT and HRT was calculated as the mean of the three measurements.
Startle paradigm The prepulse inhibition experiments were performed in four startle chambers (San Diego Instru-ments, San Diego, CA, USA). The chambers contained a plexiglass tube (diameter 8.2 cm, length 25 cm) mounted on a plastic frame, under which a piezoelectric accelerometer was attached. This device recorded and transduced the motion of the tube, which was then sent to a computer. The whole device was placed inside a sound attenuating chamber. After the rats were placed in the startle box, they were allowed to habituate to the tube and the background noise (70 dB) for 5 min. After this period they were subjected to a prepulse inhibition session. In this session, rats received 10 startle trials, 10 no-stimulus trials and 30 prepulse inhibition trials. The startle trials consisted out of a 30 ms 120 dB burst of white noise. The prepulse inhibition trials consisted of a 30 ms white-noise burst of 73, 75, or 80 dB followed, 100 ms later, by a 30 ms white noise burst of 120 dB. Each prepulse intensity was presented 10 times. During the no-stimulus condition only the background noise was presented. The 50 trials were pseudo-randomly presented with different inter-trial intervals (between 10 and 20 s). The resulting movement of the rat was recorded for 100 ms, starting at the onset of the 120 dB stimulus, with a sampling frequency of 1 kHz. Basal startle amplitude was calculated as the mean of the 10 startle trials. As there were no prepulse intensity×drugs interaction for any of the drugs tested (data not shown), we decided to only show the mean prepulse inhibition. This was calculated according to the formula PPI=100×[1–(PP73+ PP75+PP80)/3×P120], in which PP73, PP75, and PP80 are the startle response of three different prepulse trials and P120 the startle response of the startle trial.
Drugs SPD (1–16 mg/kg, obtained form the Shanghai Institute of Materia Medica, Chinese Academy of Sciences), and clozapine (5–40 mg/kg, Sigma, Zwijndrecht, the Nether-lands) were dissolved in a small amount of 1 mol/L HCl, and diluted to the appropriate concentration with saline. Finally the pH was adjusted to 4–5 by adding NaHCO3. Haloperidol (0.25–2 mg/kg, Janssen, Beerse, Belgium) was dissolved in lactic acid and diluted with saline to the appropriate concen-trations. Apomorphine (0.5 mg/kg, Brocades, ACF, the Netherlands) and MK801 (0.5 mg/kg, Sigma, Zwijndrecht, the Netherlands) were dissolved in saline. In the paw test, all drugs were injected intraperitoneally 30 min before the experiment, in analogy with previous papers, to obtain a signi-ficant reduction in motor behaviour[18]. In the prepulse inhibi-tion experiments, solvent, haloperidol, clozapine or SPD was injected intraperitoneally, 15 min before the test, in analogy with previous papers, in order to not influence the basal motor activity too much[34]. Immediately before the test, rats received a subcutaneous injection with solvent, apomorphine or MK801. The time points were chosen on the basis of our previous experiments.
Statistics As FRT and HRT give non-parametric scores, data are represented as mean±SD and drug differences were analysed with the Mann-Whitney U test. Differences in basal startle amplitude and prepulse inhibition were analysed with a one way analysis of variance (ANOVA). In addition, Swerd-low et al suggested that analysis of the raw startle data would give a better indication of a change in gating[35]. Therefore, we also analysed the raw startle data for the startle alone and the three prepulse intensities using a mixed ANOVA with prepulse intensities as within subject variable and drug treatment as between subject variable.
Results
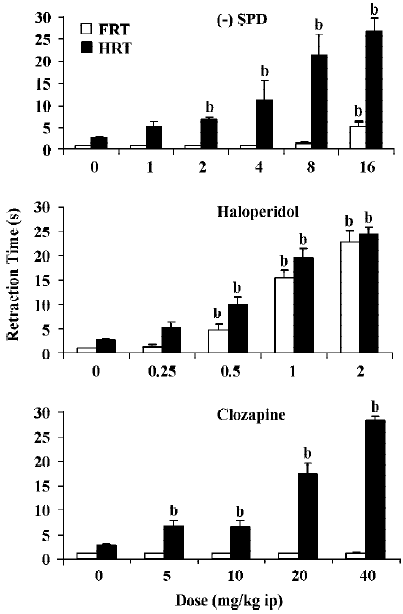
Effect of SPD, haloperidol and clozapine in the paw test All three drugs dose-dependently increased the HRT. Haloperidol also potently increased the FRT, whereas clozapine in the dose range tested did not affect FRT. SPD slightly increased FRT but only at the highest dose. Statistical analyses confirmed that SPD significantly increased HRT from a dose of 2 mg/kg. Only at 16 mg/kg did SPD significantly increase FRT. Haloperidol significantly increased both FRT and HRT at doses of 0.5 mg/kg and higher. Clozapine significantly increased HRT at all doses tested, but did not affect FRT at any of the given doses (Figure 2).
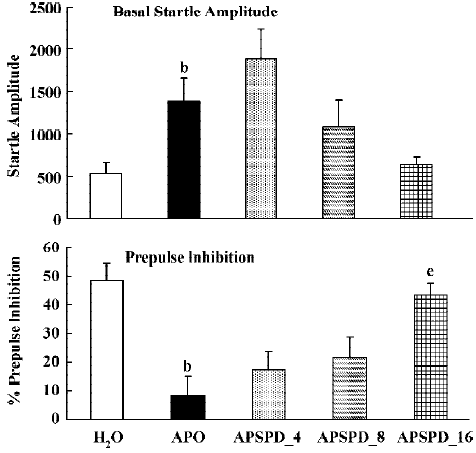
Prepulse inhibition Apomorphine significantly increased basal startle amplitude (F(1,22)<9.2; P<0.01), and significantly reduced prepulse inhibition (F(1,22)=27.8; P<0.01). Analysis of the raw startle data showed that there was a significant drug×trial interaction (F(3,66)=5.7; P<0.005). SPD did not significantly affect the basal startle amplitude of apomorphine (F(3,32)<1). However, SPD did significantly reverse the effect of apomorphine on prepulse inhibition (Figure 3B, F(3,32)= 8.2; P<0.01). Post hoc LSD analysis showed that both 8 and 16 mg/kg were significantly different from apomorphine alone. Analysis of the raw data showed a small but not significant drug × trial interaction (F(9,96)=1.85; P=0.06) (Figure 3).
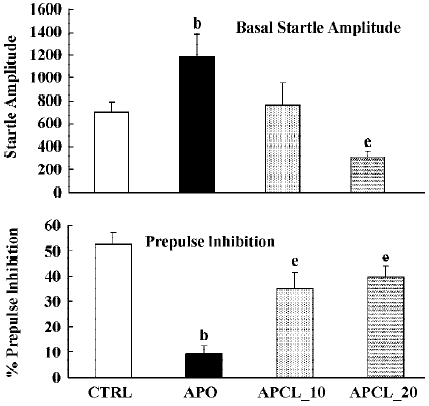
Again, apomorphine increased the basal startle response (F(1,22)=8.9; P<0.01) but decreased prepulse inhibition (F(1,22)=26.4; P<0.01). The analysis of the raw data again showed a significant drug × trial interaction (F(3,66)=6.1; P<0.01). When combined with apomorphine. clozapine significantly reversed the apomorphine-induced disruption of prepulse inhibition (F(2,25)=3.5; P<0.04), and the basal startle amplitude (F(2,25)= 6.6; P<0.01). Post hoc analysis showed that both doses of clozapine significantly reversed the effects of apomorphine on prepulse inhibition. Analysis of the raw data showed again, as in the case of SPD, that there was a nonsignificant drug×trial interaction (F(6,75)=1.8; P=0.1) (Figure 4).
Apomorphine reduced prepulse inhibition (F(1,22)=15.2; P<0.01) without affecting basal startle amplitude (F(1,22)=1.1; P>0.3). Analysis of the raw data showed a small, but significant drug×trial interaction (F(3,66)= 2.8; P<0.05). Like SPD and clozapine, haloperidol reversed the apomorphine-induced disruption of prepulse inhibition (F(2,25)=14.9; P<0.001). Post-hoc analysis showed that both doses of haloperidol significantly reversed effects of apomorphine on prepulse inhibition. Analysis of the raw data with haloperidol showed a significant drug×trial interaction (F(6.75)=3.6; P<0.01) (Figure 5).
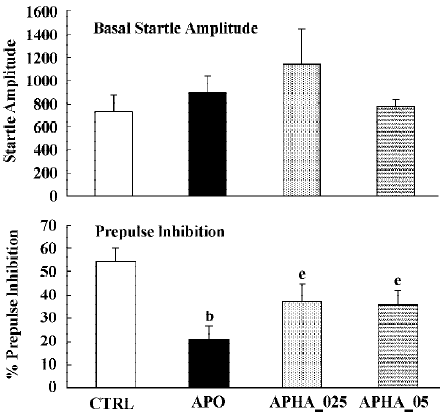
In all experiments MK801 did not affect basal startle amplitude (Figure 6: F(1,22)=3.1; P>0.09; Figure 7: F(1,22)=2.8; P>0.1; Figure 8: F(1,22)=1.1; P>0.3), but significantly reduced prepulse inhibition (Figure 6: F(1,22)=37.5; P<0.001; Figure 7: F(1,22)=34.3; P<0.01; Figure 8: F(1,22)=30.9; P<0.01). Analysis of the raw startle data showed a significant drug×trial interaction (Figure 6: F(3,66)=12.4; P<0.01; Figure 7: F(3,66)=11.6; P<0.001; Figure 8: F(3,66)=9.9; P<0.01).
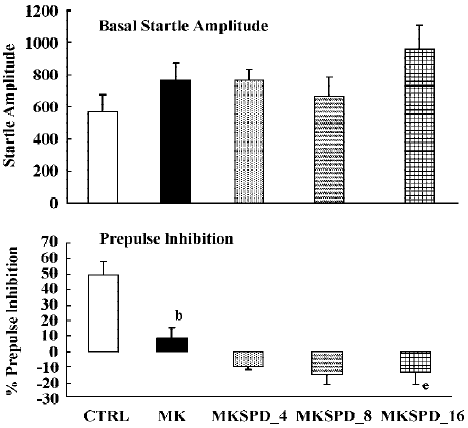
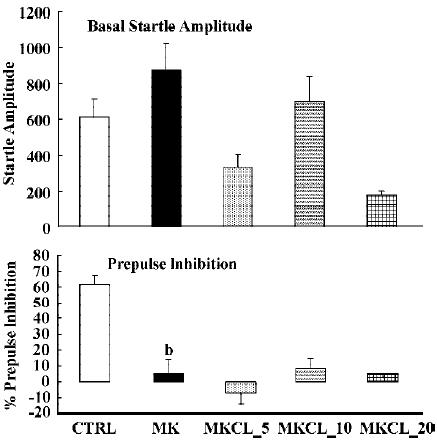
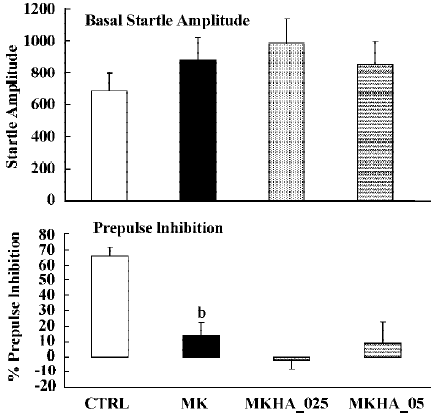
In contrast to the effects on apomorphine, neither cloza-pine (F(3,32)>1) nor haloperidol (F(2,25)=1.6; P>0.2) significantly altered this MK801-induced reduction in prepulse inhibition. Although SPD altered the effects of MK801 significantly (F(3,32)=4.1; P<0.02), inspection of Figure 6 shows that it actually potentiated the effects of MK801. Post hoc analysis showed that this effect of SPD was significant for all doses tested. Analysis of the raw startle data showed that there was a significant drug×trial interaction (F(9,96)=2.9; P<0.008).
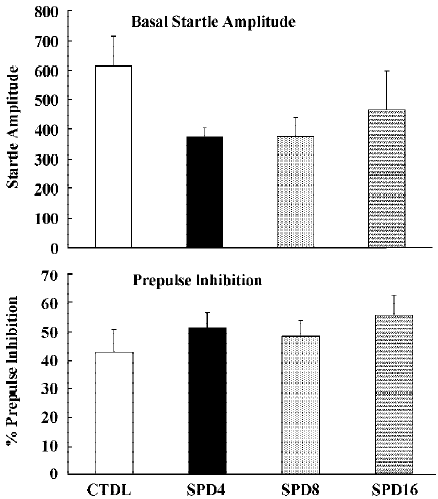
Figure 9 shows the effects of SPD when administered alone. Statistical analysis and inspection of the figure shows that SPD did not affect basal startle amplitude (F(3,31)=1.2, P> 0.35), nor prepulse inhibition (F(3,28)<1). Likewise clozapine and haloperidol did not significantly affect basal startle amplitude or prepulse inhibition (Data not shown).
Discussion
SPD represents a novel compound with D1 agonistic and D2 antagonistic dual action[17]. Because it was found to reverse dopamine agonist-induced behaviours in several paradigms, we decided to compare SPD with the standard classical antipsychotic haloperidol and the standard atypical antipsychotic clozapine in two paradigms with known validity for detecting antipsychotic drugs. The data show that SPD indeed behaved like an antipsychotic with more similarity to clozapine than to haloperidol.
In the paw test, SPD, clozapine and haloperidol dose-dependently increased the HRT. It has been shown that all antipsychotic drugs affect the HRT[19,21,36]. However, when investigating the FRT there were clear differences; where haloperidol potently increased the FRT, SPD only increased this parameter at the highest dose tested, and clozapine did not affect FRT at all. The effects of clozapine and haloperidol correspond with previous results[34,36]. The profile of SPD is similar to that of other atypical antipsychotics, including clozapine, thioridazine, olanzapine, quetiapine and risperidone[18,19,37]. At present it is difficult to explain the atypical profile of SPD in the paw test. Previous research has shown that anticholinergics and serotonin antagonists can reverse the effects on haloperidol on the forelimb retraction time, thus leading to an atypical profile like clozapine and SPD[20,38]. However, SPD has only a weak affinity for 5-HT1 or 5-HT2 receptor, and shows no affinity for muscarinic Ach receptors[39]. In contrast, SPD has D1 agonistic properties and this receptor has also been shown to influence the FRT. Although the partial agonist SKF38393 was unable to significantly reduce the effects of haloperidol on the FRT[40], a subthreshold dose of the D1 antagonist SCH39166 significantly enhanced the haloperidol induced increase in FRT[41]. These data indicate that there is a cooperative action between D1 and D2 receptors in regulating the FRT. This might explain why SPD does not influence the FRT (except at high concentrations). Its D1 agonistic properties might counteract its D2 antagonistic properties. Reasoning along these lines, at high doses the D2 antagonistic properties could be strong enough to induce a small increase in FRT. Further experiments, using a more selective full D1 agonist might shed more light on the exact nature of the D1-D2 interaction in regulating the FRT.
Apomorphine is known to strongly disrupt prepulse inhibition and it has been shown that all known antipsychotics reverse this apomorphine-induced disruption of prepulse inhibition[22,23,36]. The present study confirms that haloperidol and clozapine can dose-dependently reverse the effects of apomorphine[34]. Like these two antipsychotics, SPD also reversed the apomorphine induced disruption in prepulse inhibition (Figure 3), suggesting that SPD also has antipsychotic effects. SPD did not significantly alter the prepulse inhibition by itself, although it did tend to reduce the basal startle amplitude. This is not an uncommon finding with other D2 antagonists, although sometimes a small increase in prepulse inhibition is observed[34].
Like dopamine agonists such as apomorphine and amphetamine, glutamate antagonists, especially non-competitive antagonists such as ketamine, MK801 and phencyclidine, can reduce prepulse inhibition[27,36,42,43]. In the present study we used MK801, which also significantly reduced the prepulse inhibition. However, in contrast to the disruptive effects of apomorphine, those induced by MK801 appeared to be resistant to pretreatment with haloperidol, clozapine and SPD. The failure to reverse the MK801-induced disruption of prepulse inhibition by haloperidol is in general agreement with the published literature[22,27,36]. The situation is less clear with respect to the effects of clozapine on the glutamate antagonist-induced disruption of prepulse inhibition. As discussed in the Introduction, both reversal and non-reversal[24,26,32,33] have been described in previous studies. Our data show that in our set-up clozapine also failed to reverse the MK801-induced disruption of prepulse inhibition. Likewise, SPD did not reverse the effects of MK801. In fact, it enhanced the effects of MK801. Although the effect was only small, this might be because of a ceiling effect, as the effects of MK801 by itself were already rather strong. So far, to our knowledge, this is the first time that a (potential) antipsychotic increased the effects of MK801. It would be interesting to see whether this also applies to other behavioural effects of MK801, such as motor stimulant effects.
In summary, SPD showed the preclinical profile of an atypical antipsychotic[44]; it increased HRT, but had little influence on the FRT in the paw test, and dose-dependently reversed the apomorphine-induced disruption of prepulse inhibition. Even though it has a profile more similar to clozapine than to haloperidol there were some differences, most notably the enhancement of MK801 induced a decrease in prepulse inhibition. These data therefore suggest that SPD might be a unique novel antipsychotic. Indeed, SPD seems to be the only drug so far known in which both D1 agonist and D2 antagonist properties are combined. It has been suggested that D1 receptors, especially in the prefrontal cortex might be involved in a number of the cognitive deficits of schizophrenia, such as impaired working memory[45] and in attention[46]. Moreover, some deficits in prefrontal D1 functioning have been observed in schizophrenia[47–49] and they appear to be related to working memory deficit[50]. Unfortunately, so far, the effects of SPD in an animal model for negative symptoms or cognitive deficits have not been investigated. In addition, large-scale clinical trials will have to be performed to substantiate this claim.
References
- Jin GZ. (-)-Tetrahydropalmatine and its analogues as new dopamine receptor antagonists. Trends Pharmacol Sci 1987;8:81-2.
- Xu SX, Yu LP, Han YR, Chen Y, Jin GZ. Effects of tetrahydropro-toberberines on dopamine receptor subtypes in brain. Acta Pharmacol Sin 1989;10:104-10.
- Sun BC, Jin GZ. Characteristics of (-)-stepholidine on the firing activity of substantia nigral dopamine neurons after repeated reserpine treatment. Biol Signals 1992;1:331-8.
- Chiodo LA, Bunney BS. Typical and atypical neuroleptics: Differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J Neurosci 1983;3:1607-19.
- White FJ, Wang RY. Differential effects of classical and atypical antipsychotic drugs on A9 and A10 dopamine neurons. Science 1983;211:1054-6.
- Skarsfeldt T. Differential effects of repeated administration of novel antipsychotic drugs on the activity of midbrain dopamine neurons in the rat. Eur J Pharmacol 1995;281:289-94.
- Zhang ZD, Jin GZ, Xu SX, Yu LP, Chen Y, Jiang FY, et al. Effects of l-stepholidine on the central nervous and cardiovascular systems. Acta Pharmacol Sin 1986;7:522-6.
- Jin GZ, Huang KX, Sun BC. Dual actions of (-)-stepholidine on dopamine receptor subtypes after substantia nigra lesion. Neurochem Int 1992;20 Suppl:175S-8S.
- Zou LL, Chen Y, Song YY, Jin GZ. Effect of (-)-stepholidine on serum prolactin level of female rats. Acta Pharmacol Sin 1996;17:311-4.
- Dong ZJ, Guo X, Chen LJ, Han YF, Jin GZ. Dual action of (-)-stepholidine on the dopamine receptor mediated adenylate cyclase activity in rat corpus striatum. Life Sci 1997;61:465-72.
- Huang KX, Jin GZ. The antagonistic effects of tetrahydroproto-berberines on dopamine receptors: electrophysiological studies. Sci China B 1992;35:688-96.
- Sun BC, Jin GZ. Effects of (-)-Stepholidine on firing activity of dopamine rneurons in ventral tegmental area of the rats. Acta Pharmacol Sin 1992;13:395-9.
- Dong ZJ, Chen LJ, Jin GZ, Creese I. GTP regulation of (-)-stepholidine binding to R(H) of D1 dopamine receptors in calf striatum. Biochem Pharmacol 1997;54:227-32.
- Liu J, Guo X, Wang BC, Jin GZ. Increased phosphorylation of DARPP-32 by D1 agonistic action of l-stepholidine in the 6-OHDA-lesioned rat striatum. Acta Physiol Sin 1999;51:65-72.
- Zhu ZT, Fu Y, Hu GY, Jin GZ. Modulation of medial prefrontal cortical D1 receptors on excitatory firing activity of nucleus accumbens neurons elicited by (-)-stepholidine. Life Sci 2000;67:1265-74.
- Zhu ZT, Fu Y, Hu GY, Jin GZ. Electrophysiological study on biphasic firing activity elicted by D1 agonist-D2 antagonist action of (-)-stepholidine in nucleus accumbens. Acta Physiol Sin 2000;52:123-30.
- Jin GZ, Zhu ZT, Fu Y. (-)-Stepholidine: a potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions. Trends Pharmacol Sci 2002;23:4-7.
- Ellenbroek BA, Peeters BW, Honig WM, Cools AR. The paw test: a behavioural paradigm for differentiating between classical and atypical neuroleptic drugs. Psychopharmacology (Berl) 1987;93:343-8.
- Cools AR, Prinssen EP, Ellenbroek BA. The olfactory tubercle as a site of action of neuroleptics with an atypical profile in the paw test: effect of risperidone, prothipendyl, ORG 5222, sertindole and olanzapine. Psychopharmacology (Berl) 1995;119:428-39.
- Ellenbroek B, Cools AR. The Paw test: an animal model for neuroleptic drugs which fulfils the criteria for pharmacological isomorphism. Life Sci 1988;42:1205-13.
- Ellenbroek BA. Treatment of schizophrenia: a clinical and preclinical evaluation of neuroleptic drugs. Pharmacol Ther 1993;57:1-78.
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenic patients: a decade in review. Psychopharmacology (Berl) 2001;156:117-54.
- Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry 1994;51:139-54.
- Bast T, Zhang WN, Feldon J, White IM. Effects of MK801 and neuroleptics on prepulse inhibition: re-examination in two strains of rats. Pharmacol Biochem Behavior 2000;67:647-58.
- Feifel D, Priebe K. The effects of subchronic haloperidol on intact and dizocilpine-disrupted sensorimotor gating. Psycho-pharmacology (Berl) 1999;146:175-9.
- Johansson C, Jackson DM, Svensson L. The atypical antipsychotic, remoxipride, blocks phencyclidine-induced disruption of prepulse inhibition in the rat. Psychopharmacology (Berl) 1994;116:437-42.
- Keith VA, Mansbach RS, Geyer MA. Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol Psychiatry 1991;30:557-66.
- Varty GB, Higgins GA. Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology (Berl) 1995;122:15-26.
- Swerdlow NR, Bakshi V, Waikar M, Taaid N, Geyer MA. Seroquel, clozapine and chlorpromazine restore sensorimotor gating in ketamine-treated rats. Psychopharmacology (Berl) 1998;140:75-80.
- Yamada S, Harano M, Annoh N, Nakamura K, Tanaka M. Involvement of serotonin 2A receptors in phencyclidine-induced disruption of prepulse inhibition of the acoustic startle in rats. Biol Psychiatry 1999;46:832-8.
- Bakshi VP, Swerdlow NR, Geyer MA. Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther 1994;271:787-94.
- Varty GB, Higgins GA. Reversal of dizocilpine-induced disruption of prepulse inhibition of an acoustic startle response by the 5-HT2 receptor antagonist ketanserin. Eur J Pharmacol 1995;287:201-5.
- Wiley JL. Clozapine’s effects on phencyclidine-induced disruption of prepulse inhibition of the acoustic startle response. Pharmacol Biochem Behav 1994;49:1025-8.
- Ellenbroek BA, Liegeois JF, Bruhwyler J, Cools AR. Effects of JL13, a pyridobenzoxazepine with potential atypical antipsychotic activity, in animal models for schizophrenia. J Pharmacol Exp Ther 2001;298:386-91.
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: Current knowledge and future challenges. Psychopharmacology (Berl) 2001;156:194-215.
- Geyer MA, Ellenbroek B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuropsycho-pharmacol Biol Psychiatry 2003;27:1071-9.
- Ellenbroek BA, Lubbers LJ, Cools AR. Activity of “seroquel” (ICI 204,636) in animal models for atypical properties of antipsychotics: a comparison with clozapine. Neuropsychophar-macology 1996;15:406-16.
- Ellenbroek BA, Prinssen EP, Cools AR. The role of serotonin receptor subtypes in the behavioural effects of neuroleptic drugs. A paw test study in rats. Eur J Neurosci 1994;6:1-8.
- Jin GZ, Sun BC. (-)Stepholidine and its analogs act on brain dopamine receptors. Prog Nat Sci 1995;5:55-63.
- Ellenbroek BA, Artz MT, Cools AR. The involvement of dopamine D1 and D2 receptors in the effects of the classical neuroleptic haloperidol and the atypical neuroleptic clozapine. Eur J Pharmacol 1991;196:103-8.
- Prinssen EP, Ellenbroek BA, Stamatovic B, Cools AR. The effects of haloperidol and raclopride in the paw test are influenced similarly by SCH 39166. Eur J Pharmacol 1993;231:275-80.
- Mansbach RS. Effects of NMDA receptor ligands on sensorimotor gating in the rat. Eur J Pharmacol 1991;202:61-6.
- Mansbach RS, Geyer MA. Parametric determinants in pre-stimulus modification of acoustic startle: interaction with ketamine. Psychopharmacology (Berl) 1991;105:162-8.
- Zhang XX, Sun BC, Jin GZ. Atypical neuroleptic properties of (-)-stepholidine. Sci China B 1997;40:531-9.
- Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in the prefrontal cortex: involvement in working memory. Science 1991;251:947-50.
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into the rat prefrontal cortex. J Neurosci 2000;20:1208-15.
- Knable MB, Hyde TM, Murray AM, Herman MM, Kleinman JE. A postmortem study of frontal cortical dopamine D1 receptors in schizophrencs, psyciatric controls, and normal controls. Biol Psychiatry 1996;40:1191-9.
- Hess EJ, Bracha HS, Kleinman JE, Creese I. Dopamine receptor subype imbalance in schizophrenia. Life Sci 1987;40:1487-97.
- Albert KA, Hemmings HC Jr, Adamo AI, Potkin SG, Akbarian S, Sandman CA. Evidence for decreased DARPP-32 in the prefrontal cortex of patients with schizophrenia. Arch Gen Psychiatry 2002;59:705-12.
- Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 2002;22:3708-19.