
Gene transfer of heat-shock protein 20 protects against ischemia/reperfusion injury in rat hearts1
Introduction
Members of the small heat shock protein (sHSP) family, including HSP20, HSP25, HSP27, αB-crystallin, and myotonic dystrophy kinase binding protein, are expressed in muscle tissues and share a homologous sequence of approximately 80–100 amino acids at the C-terminus, known as the α crystallin domain[1,2]. The past decade has witnessed the discovery of new mammalian sHSP, of which HSP20 is the best characterized. HSP20 was co-purified from skeletal muscle with αB-crystallin and HSP27 by affinity chromatography on a column of immobilized antibodies against αB-crystallin[3]. Exposure of rat diaphragm tissue to heat stress in vitro results in the redistribution of HSP20, as well as αB-crystallin and HSP27, from the cytosol into insoluble fractions, and enhanced dissociation of the aggregated form to the small form, which is characteristic of stress proteins[4]. Stable overexpression of HSP20 in Chinese hamster ovary cells results in enhanced survival after heat shock, which is similar to results obtained with αB-crystallin[5]. Chu et al[6] were the first to identify the de novo phosphorylation of cardiac HSP20 in mouse cardiomyocytes after prolonged activation of the β-adrenergic signaling pathway. The adenovirus-mediated overexpression of HSP20 in adult rat cardiomyocytes increases cell contractility, which indicates that HSP20 is involved in the regulation of myocardial contractility[6,7].
Heat shock proteins have been implicated in modulating the cellular response to many stressors, and as molecular chaperones in suppressing the aggregation or assisting in the refolding of partially denatured proteins. They usually protect against ischemic/reperfusion (I/R) injury in vitro[8–11] and in vivo[12–14]. However, whether gene transfer of the HSP20 gene into the beating heart produces a myocardial protective effect has not been shown. In the present study, we transferred the HSP20 gene through a recombinant adeno-virus encoding HSP20 into the myocardium, and showed that HSP20 protected against I/R injury, probably by reducing myocardial apoptosis and necrosis in rats.
Materials and methods
Animals and experimental protocols Male adult Sprague-Dawley rats (230 g–280 g) received a standard diet and free water. The treatment of the animals and experimental protocols adhered to the guidelines of the Health Sciences Center of Peking University (Beijing, China). The animals were allowed to readjust to the new housing environment for 1 week before the experiments.
Rats were assigned randomly to 4 groups. In the no-vector control group, the chest was opened and injected with saline. The I/R control group was also injected with saline. The third group received the recombinant adenovirus encoding wild-type HSP20 (Ad.HSP20), and the fourth group received the recombinant adenovirus encoding green fluorescent protein (Ad.GFP).
Construction of recombinant adenoviruses The recombinant Ad.HSP20 and Ad.GFP were prepared as described previously[15]. Adenovirus was propagated in 293 cells and purified by 2 rounds of CsCl density ultracentrifugation (4 °C, 13 000×g for 105 min and 16 h, respectively). Viral stocks were then desalted through a PD-10 desalting column (Amersham Biosciences, Buckinghamshire, UK) into a Tris-buffered solution (10 mmol/L Tris, pH 8.0, 2 mmol/L MgCl2 and 4% sucrose)[16], plaque-titered, aliquoted, and stored at -80 °C with 4% sucrose until use.
In vivo intracoronary delivery of adenoviruses The surgical procedures were carried out as described previously[17]. Donor rats were anesthetized with sodium pentobarbital (50 mg/kg, ip). Further injections were given as needed throughout the surgical procedure. Animals were placed supine on a thermoregulated table (37 °C) . The surgery was carried out under sterile conditions. The animals were intubated and ventilated on a positive-pressure ventilator. The tidal volume was set at 1.5 mL–2.5 mL, and the respiratory rate was adjusted to within the range of 80 cycles/min to 90 cycles/min to maintain normal arterial paO2, paCO2, and pH. The chest was entered through a left intercostal approach. Before virus infusion, adenosine (0.15 mg), lidocaine (0.03 mg), and heparin (50 U) were administered via the jugular vein. With the use of a 26 gauge needle, 200 µL diluted replication-deficient adenovirus (2.2×1010 pfu) or 200 µL sterile saline were injected from the apex of the left ventricle into the left ventricular cavity while the aorta and pulmonary arteries were clamped just above the aorta root. The clamp was maintained for 15 s when the heart pumped against a closed system. After injection, the exposed heart was monitored for 5 min for resumption of normal sinus rhythm. Hemodynamic indices were measured and electrocardiography was carried out throughout the experimental period.
Myocardial infarction protocol Four days after the injection of saline or virus, the animals were re-anesthetized and ventilated artificially with room air. The thorax was reopened and the heart were exposed to identify the left anterior descending coronary artery (LAD). A 7-0 silk suture was passed around the LAD with an atraumatic needle just 4 mm inferior to the left auricle, and the artery was occluded by snaring with a vinyl tube through which the ligature had been passed. The coronary artery was occluded by pulling the snare tight and securing it with a hemostat. Ischemia was confirmed by myocardial blanching and electrocardiography evidence of injury. After 20-min ischemia, the ligature was released and the heart was reperfused for 2 h. Reperfusion was identified by an obvious ST segment change.
Measurement of infarction At the end of the infarction protocol, the ligature around the LAD was retightened and 0.1 mL of 10% Evans blue dye was injected as a bolus into the left ventricle (LV) cavity with a 26-gauge needle positioned in the apex of the heart. When the eyes turned blue, the animals were euthanized immediately, the heart was excised and rinsed in water to remove excess dye, the atria and right ventricular free wall were removed, and the remaining LV was frozen. The LV was then cut from apex to base into 4−6 transverse slices of 2 mm thick. Each slice was weighed and then incubated in 4% triphenyltetrazolium chloride solution (TTC) in isotonic pH 7.4 phosphate buffer at 37 °C for 30 min. The slices were subsequently fixed in 10% formalin solution for 24 h. Viable tissue (red-stained by the TTC) was distinguished easily from the infarcted regions (pale or unstained by the TTC) and the risk area (unstained by Evans blue). The total slice area, the infarcted area, and the risk area of each slice were determined by computer-assisted planimetry (Leica Qwin image analysis software; Leica, Cambridge, UK). During planimetry, the operator was blinded as to the type of animal. The ratios of risk area to total slice area, infarct area to total slice area, and infarct area to risk area were calculated and multiplied by the weight of the slice to determine risk and infarct weight per slice. Infarct size was expressed as a proportion of LV mass or risk area mass.
Hemodynamic studies Hemodynamic measurements were taken at 0 min, 10 min and 20 min ischemia and 30 min, 60 min and 120 min reperfusion. A 1.5 F micronanometer-tipped catheter was advanced into the LV through the right carotid artery. The heart rate, blood pressure, left ventricular end diastolic pressure (LVEDP), left ventricular end systolic pressure (LVESP) and maximal rates of pressure increase (+dp/dtmax) and decrease (-dp/dtmax) were recorded on a polygraph (NEC San-ei Instruments, Japan).
Measurement of serum cardiac troponin T (cTnT) and creatine phosphokinase (CK) levels At the end of the myocardial I/R experiment, a 1-mL blood sample was obtained from the carotid cannula, stored at 4 °C for 30 min, and centrifuged at 3000×g for 10 min. The serum was stored at -40 °C prior to analysis. The concentration of serum cTnT was determined by the short-turn-around-time (STAT) assay (Roche Diagnostics, Basel, Switzerland), with use of the Roche Elecsys 2010 immunoassay analyzer (Roche Diagnostics). The serum was also analyzed spectrophotometrically for CK activity (Roche Diagnostics).
Western blot analysis Three hearts from each of the experimental groups were used separately for measurement of HSP20 by Western blot analysis. After 4 d of injection of saline or virus, the hearts were removed quickly, and the LV was separated and frozen in liquid nitrogen. The frozen LV tissue was homogenized in protein extraction buffer containing 20 mmol/L Tris-HCl, pH 7.4, 1% Trion X-100, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetracetic acid, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L NaF, 1 mmol/L Na3VO4 and 0.1 mmol/L phenylmethylsulfonyl fluoride. Aliquots were resolved on sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes (Schleicher & Schuell, Keene, NH, USA) and incubated with primary polyclonal anti-HSP20 antibodies (1:1000) (presented by Prof Rui-ping XIAO, NIH, USA), which recognized HSP20, at 4 °C overnight. Bound antibodies were detected using a secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Inc, CA,USA) and visualized by use of an enhanced chemiluminescence kit (SuperSignal® West Pico Trial Kit, Pierce Biotechnology, Inc, IL, USA) and exposed to X-ray film for the appropriate time.
Terminal dUTP nick-end labelling staining Hearts were isolated from each group after I/R for analysis using the terminal dUTP nick-end labeling (TUNEL) assay. Tissue samples were fixed in a 4% paraformaldehyde solution, paraffin embedded, and cut transversely into 6-µm sections. The assay was operated according to the manufacturer’s instructions (DeadEnd™ Fluorometric TUNEL System; Promega, WI, USA). Stained samples were analyzed using a confocal microscope; at least 500 cells were counted in randomly selected views.
Statistical analysis Data were expressed as mean±SD. Differences were analyzed for significance by one-way repeated-measures ANOVA and further analyzed with the use of the Newman-Keuls test for multiple comparisons between treatment groups. The results were considered significant at P<0.05.
Results
Expression of HSP20 Intraventricular injection of Ad.HSP20 in vivo resulted in increased HSP20 expression in the LV as compared with vector and Ad.GFP treatments (Figure 1A). The hearts treated with Ad.GFP showed only low-level of HSP20 expression, which indicates that treatment with viral vectors has no significant effect on HSP20 expression in the rat myocardium. LV treated with Ad.HSP20 showed homogenous expression of GFP, whereas those treated with vector showed no background fluorescence (Figure 1B).

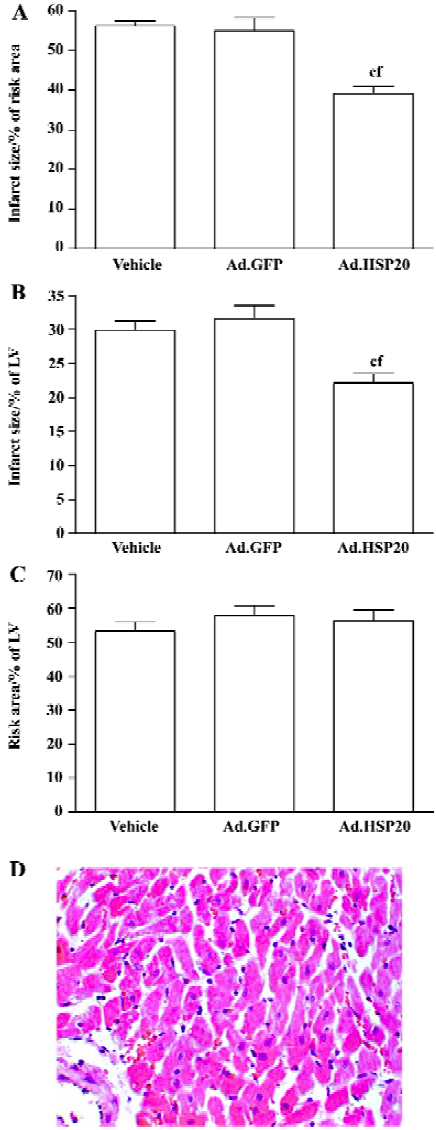
Myocardial infarction Ad.HSP20-treated hearts showed a significant reduction in infarct size (39.2%±4.3% risk area) compared with vector- and Ad.GFP-treated hearts (56.3%±2.9% and 54.9%±8.1%, respectively; P<0.01; Figure 2A). Infarct size did not differ between vector- and Ad.GFP-treated hearts (P>0.05). A similar result was observed when infarct size was expressed as a proportion of LV (Figure 2B). Both results suggest that the reduced infarct size observed in Ad.HSP20-injected hearts is entirely due to the overexpres-sion of HSP20. However, the risk areas (% of LV) were not significantly different among the groups (ie 53.2%±6.5%, 57.9%±7.3%, and 56.4%±7.5% in the vector-, Ad.GFP-, and Ad.HSP20-treated groups, respectively; P>0.05; Figure 2C).
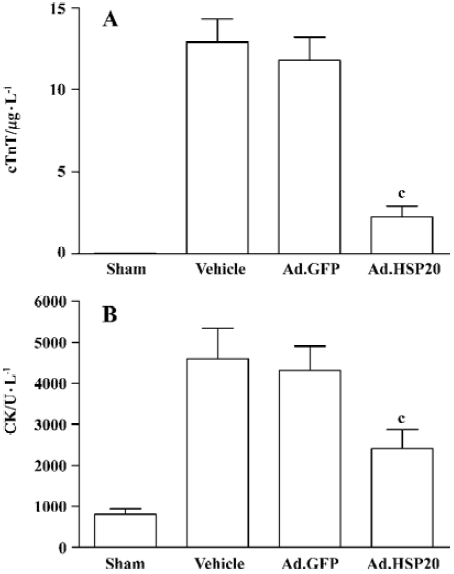
HSP20 gene delivery reduced serum cTnT and CK levels Ad.HSP20-treated hearts showed a significant reduction in cTnT release (2.2 µg/L±1.7 µg/L) compared with vector- and Ad.GFP-treated hearts (12.9 µg/L±3.2 µg/L, and 11.8 µg/L±3.1 µg/L, respectively; P<0.01; Figure 3A). Similar results were observed for CK release (Figure 3B).
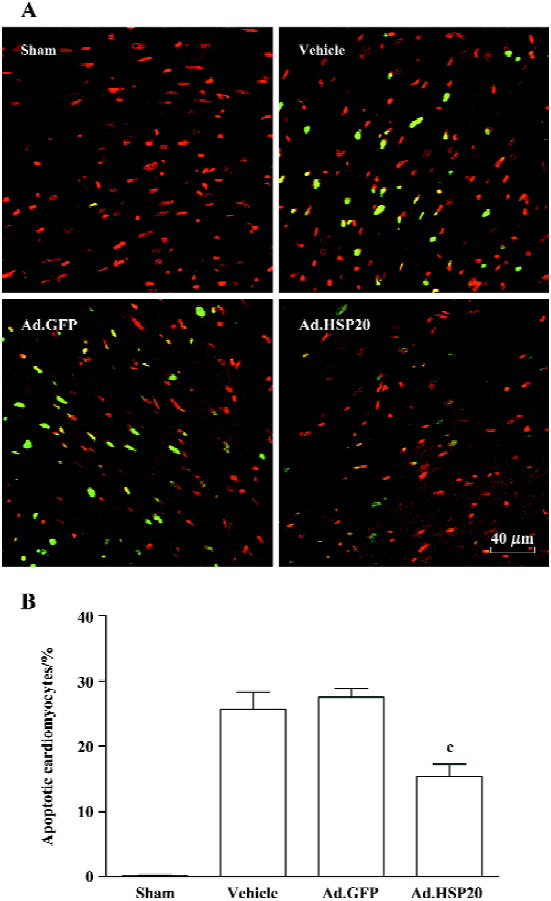
HSP20 gene delivery attenuated apoptosis in the acute ischemia/reperfusion rat model Figure 4A shows representative apoptotic cardiomyocytes identified by TUNEL staining in the I/R-injured region. The ratio of TUNEL-positive cardiomyocytes to total number of cardiomyocytes in the Ad.HSP20 group was significantly reduced as compared with the vector and Ad.GFP groups (15.4%±3.2% vs 25.7%±4.5% and 27.6%±2.2%, P<0.01; Figure 4B).
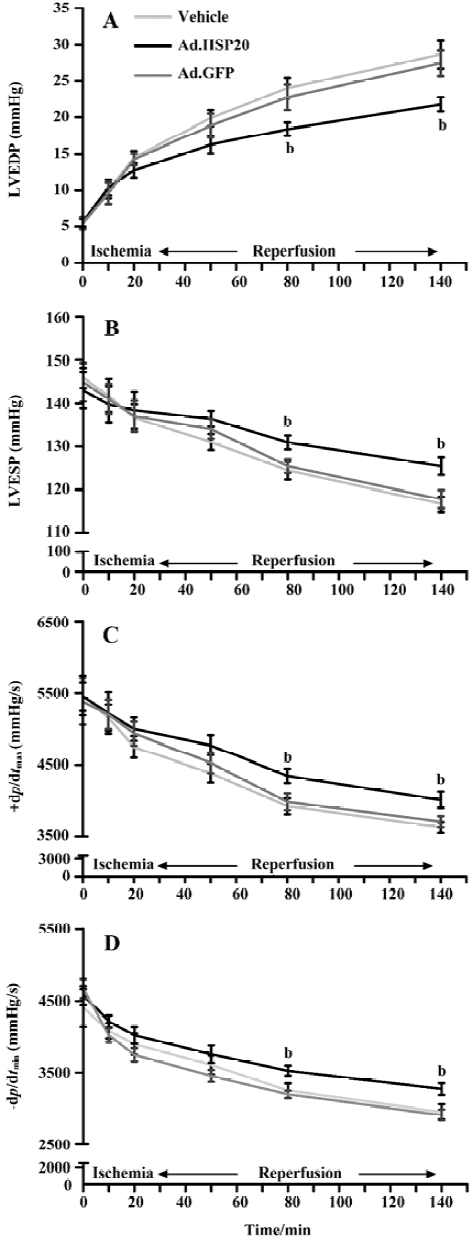
HSP20 gene delivery improved cardiac function in vivo LVEDP, LVESP, +dp/dtmax and -dp/dtmax values are shown in Figure 5. All parameters were comparable among the 3 groups before and during ischemia and during reperfusion. All parameters, except LVEDP, declined in value after ischemia. LVEDP in HSP20-treated rats was significantly decreased after 60-min reperfusion compared with that in vector- and Ad.GFP-treated rats (P<0.05, Figure 5A). LVESP, +dp/dtmax, and -dp/dtmax values in HSP20-treated rats were significantly increased after 60-min reperfusion compared with those in vector- and Ad.GFP-treated rats (P<0.05, Figure 5B, 5C, 5D).
Discussion
Gene therapy has emerged as a genuine alternative therapy in coronary artery disease, including ischemic heart disease. One of the commonly used intramyocardial gene transfer methods is direct intramyocardial injection. There have been a few promising trials involving the use of direct intramyocardial injection in this area[18,19]. However, the technical problems with this method are that only a small volume of the myocardium is accessible for transfection and the distribution of transgenes in the myocardium is not homo-geneous. To overcome these problems, we used an in vivo intracoronary gene delivery method that modified the approach of Hajjar et al[17] to transduce the HSP20 gene into the ventricular muscle with the use of recombinant adenoviral vectors. The adenoviral vectors are delivered into the myocardium via the coronary circulation. Using this delivery method, we sought to elucidate a direct cause and effect relationship between HSP20 and cardioprotective effects in the intact rat heart.
The present study demonstrates, for the first time, that gene transfer of Ad.HSP20 into the LV muscle causes robust expression of HSP20 as compared with vector or Ad.GFP transfer. Ad.GFP-treated LV showed no significant increase in HSP20 expression compared with vector-treated LV. These results suggest that the increased expression in the Ad.HSP20-treated hearts is not due to virus-related stress.
Infarct size was reduced significantly in the I/R hearts injected with Ad.HSP20. We also examined serum cTnT and CK levels independently. cTnT originating exclusively from the myocardium clearly differs from skeletal muscle troponin T. As a result of its high tissue specificity, cTnT is a cardio-specific, highly sensitive marker for myocardial damage[20]. Our results showed that the cTnT level in Ad.HSP20-treated hearts was reduced significantly as compared with that in vector- and Ad.GFP-treated hearts. Similar results were shown with CK. TUNEL staining showed that apoptosis of cardiomyocytes was reduced in Ad.HSP20-treated hearts.
The decrease of LVEDP and increase of LVESP, +dp/dtmax and -dp/dtmax in HSP20-treated hearts may be explained by HSP20 being an actin-associated protein. It is biochemically associated with αB-crystallin and localized to distinct transverse bands in a pattern similar to αB-crystallin and sarcomeric actin[21,22]. Phosphorylated HSP20 increases the contractility rate of cardiac myocytes, which indicates that HSP20 is involved in the regulation of myocardial contractility[6,7].
Heat shock proteins are a family of endogenous protective proteins. Various HSP have protective effects against stress injury. HSP70 prevents cell death by inhibiting apoptosis via associating with apoptosis protease activating factor-1 (Apaf-1) and blocking the assembly of a functional apoptosome[23]. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiomyocytes protects mitochondrial function and decreases apoptotic cell death induced by simulated I/R accompanied by decreased mitochondrial cytochrome c release and caspase-3 activity[11]. HSP60 interacts with Bax and Bak to regulate apoptosis[24]. Overexpression of αB-crystallin in transgenic mice hearts provides resistance to I/R injury by negatively regulating myocyte and non-myocyte apoptosis[25]. HSP27 binds to cytochrome c released from the mitochondria into the cytosol and prevents cytochrome c-mediated interaction of Apaf-1 with procaspase-9[26]. These results highlight the notion that the protective effects of HSP are closely related to mitochondrial function. Thus, HSP are anti-apoptotic proteins in cardiomyocytes.
Myocardial ischemia is followed frequently by reperfusion. Reperfusion and the resultant re-oxygenation lead to the generation of oxygen radicals that can cause reperfusion injury. Our results in vivo are consistent with those of Fan et al[15], who showed that HSP20 and its phosphorylation at Ser16 might provide protective effects against β-agonist-induced apoptosis in vitro.
Death of cardiomyocytes due to I/R injury is caused by 2 distinct mechanisms, necrosis, and apoptosis, which contribute independently to myocardial infarction[25,27]. The infarct area represents cell death, including necrotic cell death and apoptotic cell death. cTnT and CK are indicators of myocardial necrosis, whereas TUNEL staining can reveal apoptosis. Thus, our results suggest that the protective effect of HSP20 is attributed to a reduction of necrosis and apoptosis in cardiomyocytes. In addition, our recent data shows that lactate dehydrogenase release and caspase-3 activity in H9c2 cells infected with Ad.HSP20 are also decreased. Therefore, the cardioprotective effect of HSP20 in vivo might be mediated mainly by inhibiting both cardio-myocyte necrosis and apoptosis.
In conclusion, our results show that overexpression of HSP20 protects against I/R injury in vivo, not only by inhibi-ting cardiomyocyte necrosis and apoptosis but also by increasing myocardial contractility. Our data suggest that HSP20 is a potential therapeutic protein for ischemic diseases and additional experiments should be carried out.
Acknowledgements
We are grateful to Prof Rui-ping XIAO (Laboratory of Cardiovascular Science, Gerontology Research Center, National Institutes of Health, Bethesda, MD, USA) for her advice during this research.
References
- Boelens WC, Croes Y, de Ruwe M, de Reu L, de Jong WW. Negative charges in the C-terminal domain stabilize the αB-crystallin complex. J Biol Chem 1998;273:28085-90.
- Suzuki A, Sugiyama Y, Hayashi Y, Nyu IN, Yoshida M, Nonaka I, et al. A novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J Cell Biol 1998;140:1113-24.
- Kato K, Shinohara H, Goto S, Inaguma Y, Morishita R, Asan T. Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. J Biol Chem 1992;267:7718-25.
- Kato K, Goto S, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20-kDa protein that is highly homologous to αB crystallin. J Biol Chem 1994;269:15302-9.
- van de Klundert FAJM, van den IJssel PRLA, Stege GJ, de Jong WW. Rat Hsp20 confers thermoresistance in a clonal survival assay, but fails to protect coexpressed luciferase in Chinese hamster ovary cells. Biochem Biophys Res Commun 1999;254:164-8.
- Chu G, Egnaczyk GF, Zhao W, Jo SH, Fan GC, Maggio JE, et al. Phosphoproteome analysis of cardiomyocytes subjected to β-adrenergic stimulation: identification and characterization of a cardiac heat shock protein p20. Circ Res 2004;94:184-93.
- Pipkin W, Johnson JA, Creazzo TL, Burch J, Komalavilas P, Brophy C. Localization, macromolecular associations, and function of the small heat shock-related protein HSP20 in rat heart. Circulation 2003;107:469-76.
- Mestril R, Chi SH, Sayen MR, O’Reilly K, Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest 1994;93:759-67.
- Lau S, Patnaik N, Sayen MR, Mestril R. Simultaneous over-expression of two stress proteins in rat cardiomyocytes and myogenic cells confers protection against ischemia-induced injury. Circulation 1997;96:2287-94.
- Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation 1997;96:4343-8.
- Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, Dillmann WH. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation 2001;103:1787-92.
- Okubo S, Wildner O, Shah MR, Chelliah JC, Hess ML, Kukreja RC. Gene transfer of heat-shock protein 70 reduces infarct size in vivo after ischemia/reperfusion in the rabbit heart. Circulation 2001;103:877-81.
- Hutter JJ, Mestril R, Tam EK, Sievers RE, Dillmann WH, Wolfe CL. Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation 1996;94:1408-11.
- Zhong N, Zhang Y, Fang QZ, Zhou ZN. Intermittent hypoxia exposure-induced heat-shock protein 70 expression increases resistance of rat heart to ischemic injury. Acta Pharmacol Sin 2000;21:467-72.
- Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ Res 2004;94:1474-82.
- Nyberg-Hoffman C, Aguilar-Cordova E. Instability of adenoviral vectors during transport and its implication for clinical studies. Nat Med 1999;5:955-7.
- Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci USA 1998;95:5251-6.
- Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, et al. Gene therapy for myocardial angiogenesis. Initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation 1998;98:2800-4.
- Rosengart TK, Lee LY, Patel SR, Sanborn TA, Parikh M, Bergman GW, et al. Angiogenesis gene therapy: phase I assessment of direct intramyocardial administration of an adenovirus vector expression VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation 1999;100:468-74.
- Zacharowski K, Otto M, Hafner G, Chatterjee PK, Thiemermann C. Endotoxin induces a second window of protection in the rat heart as determined by using p-nitro-blue tetrazolium staining, cardiac troponin T release, and histology Arterioscler Thromb Vasc Biol 1999;19:2276-80.
- Brophy CM, Lamb S, Graham A. The small heat shock-related protein-20 is an actin-associated protein. J Vasc Surg 1999;29:326-33.
- Rembold CM, Zhang E. Localization of heat shock protein 20 in swine carotid artery. BMC Physiol 2001;1:10.
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000;2:469-75.
- Kirchhoff SR, Gupta S, Knowlton AA. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation 2002;105:2899-904.
- Ray PS, Martin JL, Swanson EA, Otani H, Dillman WH, Das DK. Transgene overexpression of αB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J 2001;15:393-402.
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, et al. Hsp27 negatively regulates cell death by interacting with cytochrome C. Nat Cell Biol 2000;2:645-52.
- Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, et al. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest 1996;74:86-107.
- Zhu YH, Wang X. Overexpression of heat-shock protein 20 in rat heart myogenic cells confers protection against simulated ischemia/reperfusion injury. Acta Pharmacol Sin 2005;26:1076-80.