
Suberoyl bis-hydroxamic acid induces p53-dependent apoptosis of MCF-7 breast cancer cells1
Introduction
Suberoyl bis-hydroxamic acid (SBHA), a relatively new histone deacetylase (HDAC) inhibitor, has been the subject of studies on cell division[1] and fetal globin gene expression[2]. SBHA can induce apoptosis in the majority of melanoma cell lines through a mitochondrial and caspase-dependent pathway[3]. Several key anti-apoptotic proteins, including X-linked inhibitor of apoptosis protein (IAP) and Bcl-2 family proteins, Mcl-1 and Bcl-XL, are downregulated in SBHA-treated cells. In contrast, SBHA induces the upregulation of Bcl-2 family pro-apoptotic proteins Bid, Bim, Bax, and Bak. Moreover, the expression levels of procaspases-8 and -3 are also upregulated by SBHA treatment[4]. So far, however, there are no reports as to whether SBHA can induce apoptosis in MCF-7 cancer cells and how this affects the p53-mediated apoptotic signaling pathway.
In the present study, we showed that HDAC inhibitor SBHA induced apoptosis in MCF-7 breast cancer cells, thus demonstrating the cytotoxic effects of SBHA. We also showed that SBHA increased the expressions of p53, p21, Bax, and PUMA during apoptosis. However, the expressions of p21, Bax, and PUMA decreased when p53 was inhibited by p53 siRNA, and at the same time, SBHA-induced apoptosis was blocked. These results showed that SBHA might induce cytotoxicity in MCF-7 cells via the activation of p53-mediated apoptotic signaling.
Materials and methods
Cell culture and drug preparation Human MCF-7 breast cancer cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum in a humidified atmosphere with 5% CO2 at 37 ºC. For the experiment, the MCF-7 cells were incubated with 40 µmol/L SBHA (Calbiochem, Gibbstown, NJ, USA) or RPMI-1640 medium alone for 24 h. In total, 8.168 mg SBHA was dissolved in 0.8168 mL DMSO, and this solution was then dissolved in 1 L medium. The final concentration of SBHA was 40 µmol/L, and the final concentration of DMSO was 0.08168% (<0.5%). The same concentration of DMSO was used in the control experiments.
Morphological observation of apoptotic cells After exposure to the indicated conditions, the MCF-7 cells were centrifuged and washed with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde, and stained with Hoechst 33258 (5 µg/mL; Sigma-Aldrich, St Louis, MO, USA) for 15 min at room temperature. Apoptosis was detected by fluorescent microscopy. The results are representative of 6 similar experiments.
Flow cytometric analysis A total of 1×106 MCF-7 cells were harvested after SBHA treatment for 48 h. The cells were washed twice with cold PBS, resuspended in 2 mL of 70% ethanol, and kept at 4 ºC overnight. They were subsequently rinsed twice with PBS and incubated with 100 µL RNase (10 mg/mL). Finally, the cells were stained with BrdU or propidium iodide. Distribution of the cell cycle and the rate of apoptosis were determined by flow cytometry (Becton Dickinson, Franklin Lakes, USA)[5].
Detection of DNA fragmentation The total cellular DNA was extracted from the cells. In brief, the cells were washed and lysed overnight at 37 ºC in lysis buffer containing 10 mmol/L Tris-HCl (pH 8.0), 10 mmol/L edetic acid, 0.4% sodium dodecylsulfate, and 100 mg/L proteinase K. Subsequently, the cells were treated with 0.5 µg/mL RNase A for 2 h. The genomic DNA was extracted by phenol-chloroform isoamyl alcohol extraction. Electrophoresis was performed on 2% agarose gel. The DNA was visualized by UV illumination.
RT-PCR Total RNA was extracted from the cells with TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). The reverse transcription reaction was performed using the Superscript first-strand synthesis system (Invitrogen Life Technologies, USA). The newly-synthesized cDNA was amplified by PCR. Human P53 (forward, 5'-ACTAAGCGAGCACTGCCCAAC-3'; reverse, 5'-CCTCATTCAGCTCTCGGAACATC-3'), PUMA (forward, 5'-CGACCTCAACGCACAGTACGA-3'; reverse, 5'-GGCACCTAATTGGGCTCCATC-3'), p21 (forward, 5'-CTGGGGATGTCCGTCAGAAC-3'; reverse, 5'-GAGTCTCCAGGTCCACCTGG-3'), and Bax primers (forward, 5'-TGGCAGCTGACATGTTTTCTGAC-3'; reverse, 5'-TCACCCAACCACCCTGGTCTT-3'). GAPDH (5'-GCCAAAAGGGTCATCATCTC-3'; 5'-GTAGAGGCAGGGATGATGTTC-3') was used as an internal control. Amplication cycles were: 94 °C for 3 min, then 33 cycles at 94 °C for 1 min, 56 °C for 1 min, 72 °C for 1.5 min, followed by 72 °C for 10 min. Aliquots of PCR product were electrophoresed on 1.5% agarose gels, and PCR fragments were visualized by ethidium bromide staining. mRNA expression was quantified by Molecular Analyst software Image.
Western blot analysis To prepare the whole-cell extract, cells were washed, lysed in cold TNT buffer (20 mmol/L Tris-HCl [pH 7.4], 200 mmol/L NaCl, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, and 1% aprotinin) for 45 min. The lysates were centrifuged at 12 000×g for 40 min at 4 °C. The supernatants (50 µg protein) were resolved by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane, which was incubated in blocking solution consisting of 5% powered milk in TBST (10 mmol/L Tris-HCl [pH 8.0], 150 mmol/L NaCl, and 0.1% Tween 20) for 1 h, then immunoblotted with an anti-p53 antibody (sc-99), anti-PUMA antibody (sc-19187), anti-p21 antibody (sc-817), and anti-Bax antibody (sc-6236; (Santa Cruz Biotechnology, Santa Cruz, CA, USA; the dilution of each antibody was 1:500) or antitubulin antibody (T6074; Sigma-Aldrich, USA; dilution: 1:2000). Detection by enzyme-linked chemiluminescence was performed according to the manufacturer’s protocol (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Protein expression was quantified by Molecular Analyst software ImageJ.
Determination of mitochondrial membrane potential JC-1 staining was performed according to the manufacture’s instructions (Molecular Probes, Eugene, OR, USA). Briefly, the cells were collected, washed with PBS, and then incubated with 10 μg/mL JC-1 in warm PBS for 15 min. After washing with PBS, the cells were analyzed with flow cytometry (Becton Dickinson, USA). Red and green fluorescence were expressed as percentages of total gated cells.
siRNA preparation and transfection The cells in the exponential phase of growth were seeded in 6-well plates at a concentration of 5×105 cells/well. After incubation for 24 h, the cells were transfected with siRNA for p53 (100 nmol/L; catalog N
Quantification of apoptosis The cells were stained with Hoechst 33258 and then observed under a fluorescent microscope. Apoptotic cells with condensed or fragmented nuclei were easily distinguished from normal cells with intact nuclei. The quantification of apoptosis was determined by counting the number of apoptotic cells: 6 randomly-chosen fields of view were observed after exposure to the conditions indicated, with a minimum number of 500 cells scored in each condition.
Statistical analysis Data shown represent mean±SD. Statistical analyses for the detection of significant differences between the control and experimental groups were carried out using Student’s t-test.
Results
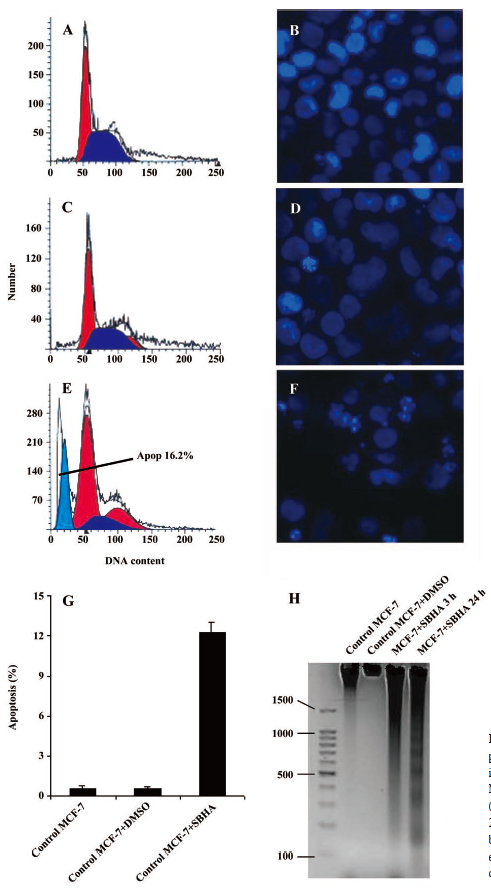
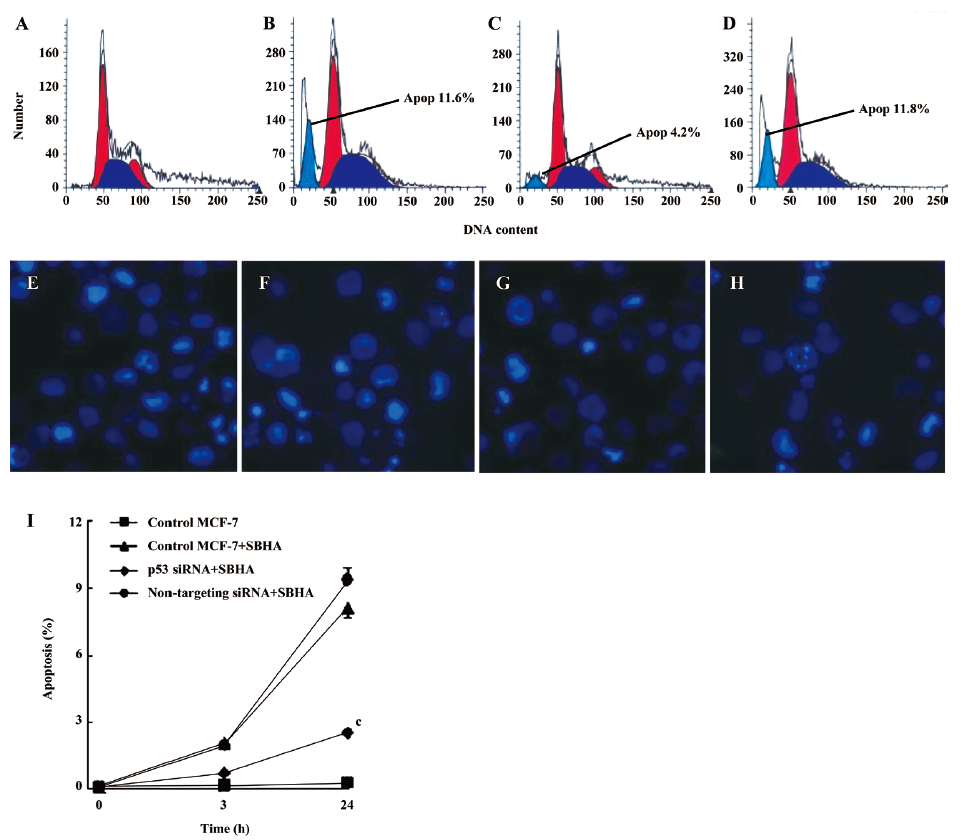
SBHA induced apoptosis in MCF-7 cells Apoptosis induced by SBHA in MCF-7 cells was examined. The cell cycle results indicated that SBHA altered the cell cycle of MCF-7 cells, showing a typical subdiploid apoptotic peak before the G1 phase after SBHA treatment for 24 h (Figure 1E). As shown with Hoechst 33258 staining, the control cells did not show signs of chromatin condensation (Figure 1B). In contrast, a dense and thin crown of nuclear coloration, typical of chromatin condensation, could be observed in the SBHA-treated cells at 24 h (Figure 1F). Although several nuclei still displayed a normal morphology, we found that most of the cells showed very intense staining of condensed and fragmented chromatin. This evidence clearly showed that SBHA induced apoptosis in MCF-7 cells. DNA agarose gel electrophoresis showed that the MCF-7 cells presented typical DNA ladder patterns of apoptosis after treatment with 40 µmol/L SBHA for 24 h (Figure 1H).
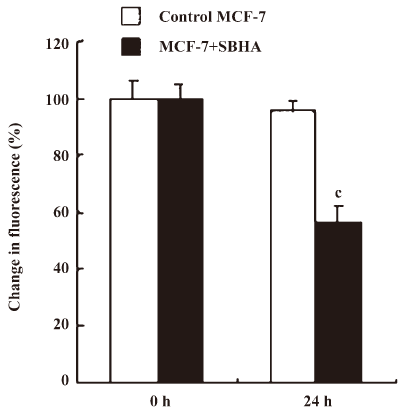
SBHA induced mitochondrial dysfunction In the present study, mitochondrial membrane potential (ΔΨm) was examined using fluorescent dye JC-1. We detected a collapse in ΔΨm at 24 h after SBHA treatment (Figure 2). A collapse in ΔΨm always indicates cell apoptosis or necrosis. SBHA induced mitochondrial dysfunction, which also proved that SBHA induced apoptosis in MCF-7 cells.
SBHA upregulated the p53 level To determine whether SBHA-induced apoptosis involved the p53 signaling pathway, RT-PCR analysis was used to measure p53 mRNA expression. The basal levels of p53 mRNA in MCF-7 cells were relatively low. Twenty-hours after incubation with SBHA, p53 mRNA expression was significantly increased (Figure 3A). To determine whether SBHA also increased the expression of the p53 protein, Western blot analysis was used. The level of the p53 protein increased at 24 h after SBHA treatment. These results showed that p53 expression was induced by SBHA treatment (Figure 4A).
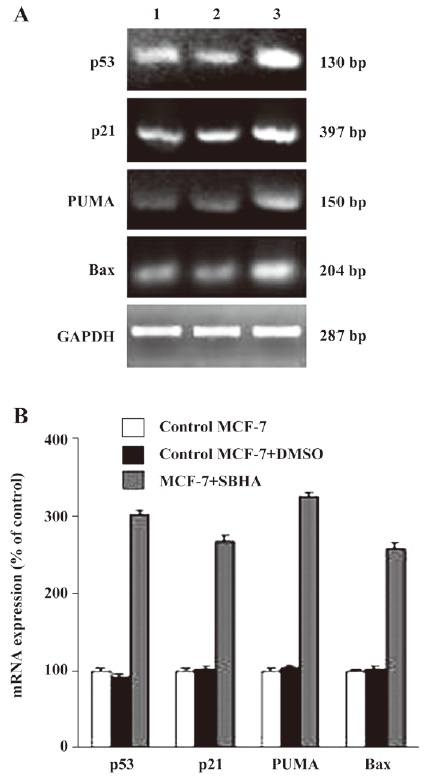
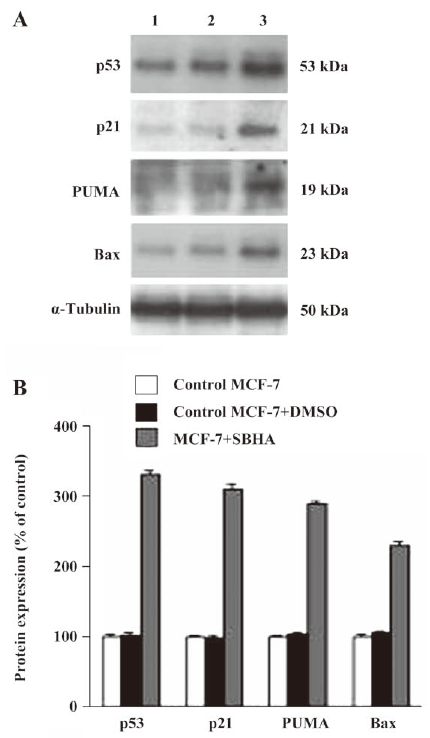
SBHA upregulated PUMA, Bax, and p21 levels To determine whether SBHA increased the expression of p53 target genes PUMA, Bax, and p21, RT-PCR analysis was used to measure PUMA, Bax, and p21 mRNA expressions. The levels of PUMA, Bax, and p21 mRNA in MCF-7 cells significantly increased after incubation with SBHA for 24 h (Figure 3A). To determine whether SBHA also increased the expressions of the PUMA, Bax, and p21 proteins, Western blot analysis was used. The PUMA, Bax, and p21 protein level significantly increased after incubation with SBHA for 24 h. These results showed that PUMA, Bax, and p21 expressions were induced by SBHA treatment (Figure 4A).
Inhibition of p53 by siRNA attenuated the SBHA-induced expression of PUMA, Bax, and p21 The MCF-7 cells were transfected with siRNA targeting p53 for 24 h, and then treated with 40 µmol/L SBHA for 24 h. As shown in Figure 5A, p53 siRNA significantly inhibited the p53 protein after SBHA treatment for 48 h. As shown in Figure 5C and 5E, p53 mRNA or protein was inhibited by p53 siRNA during SBHA treatment, and at the same time, PUMA, Bax, and p21 mRNA and proteins were also inhibited by p53 siRNA during SBHA treatment (Figure 5C, 5E).
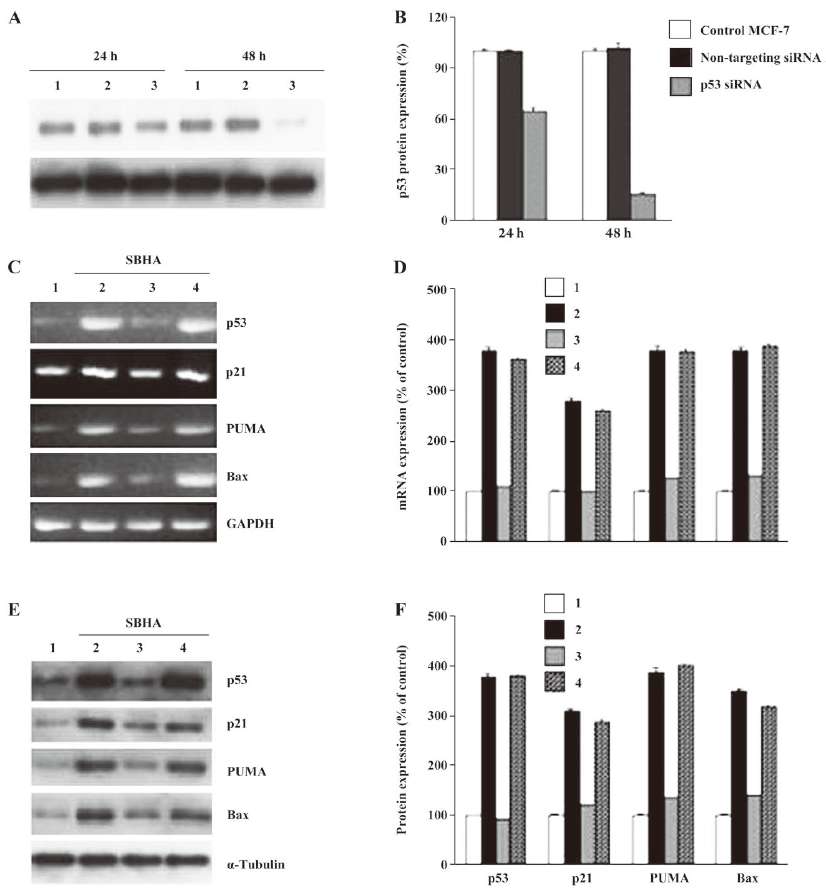
Inhibition of p53 by siRNA attenuated SBHA-induced apoptosis To further examine the role of p53 in apoptosis induced by SBHA, the MCF-7 cells were transfected with siRNA targeting P53 or non-targeting siRNA for 24 h, and then treated with 40 µmol/L SBHA for 24 h. The cell cycle results indicated that p53 siRNA significantly altered the cell cycle of apoptotic MCF-7 cells treated by SBHA, showing a decrease of the subdiploid apoptotic peak before the G1 phase (Figure 6C) compared with MCF-7 cells with SBHA treatment (Figure 6B) or MCF-7 cells transfected with non-targeting siRNA and with SBHA treatment (Figure 6D). As shown in Hoechst 33258 staining, the control MCF-7 cells with SBHA treatment (Figure 6F) or MCF-7 cells transfected with non-targeting siRNA and with SBHA treatment (Figure 6H) showed a dense and thin crown of nuclear coloration, typical of chromatin condensation. However, these typical apoptotic signs decreased in the cells transfected with p53 siRNA and with SBHA treatment (Figure 6G). The percentage of apoptotic cells was assessed by counting the cells stained with Hoechst 33258. As shown in Figure 6I, the inhibition of P53 by siRNA significantly decreased the sensitivity of MCF-7 cells to SBHA-induced apoptosis.
Discussion
In this report, we discussed the possible mechanisms of SBHA in inducing apoptosis in MCF-7 cells and the potential effect of p53 during SBHA-induced apoptosis. We found that the activation of the p53 pathway was involved in SBHA-induced apoptosis.
The tumor suppressor p53 plays a central role in controlling apoptosis. The ability of p53 to initiate apoptosis is vital for the proper regulation of cell proliferation in multicellular organisms[6,7]. p53 is activated by external and internal stress signals that promote its nuclear accumulation in an active form. In turn, p53 induces either viable cell growth arrest or apoptosis. The latter activity is crucial for tumor suppression. In our study, the basal level of p53 was low in MCF-7 cells, and SBHA upregulated the expression of p53. The upregulation of p53 after treatment with SBHA induced apoptotic cell death.
Mitochondria play a critical role in the regulation of various apoptotic processes, including drug-induced apoptosis[8]. p53 induces apoptosis via both target gene activation and transactivation-independent mechanisms at mitochondria[9]. In response to various forms of cellular stress, the level of p53 increases and a proportion of p53 rapidly localizes to the mitochondria[10]. In the present study, the collapse of mitochondrial ΔΨm may have been caused by the upregulation of p53 after SBHA treatment.
In this study, we have demonstrated that SBHA arrested the cell cycle. This biochemical event was possibly associated with p53 and p21. p53 displayed a key role in the G1/S checkpoint in response to DNA damage[11] as a regulator of cell cycle progression. p21 is the cyclin-dependent kinase inhibitor induced by cellular damage under the transcriptional control of p53. p21 plays an essential role in growth arrest after DNA damage, and its overexpression leads to G1 and G2 phase arrest[12,13]. Our results showed that SBHA caused cell cycle arrest in the S phase through the upregulation of p53 and p21.
p53 accumulates in the nucleus, where it activates a number of pro-apoptotic target genes[14] and induces apoptotic cell death. The p53 protein can engage apoptosis by inducing the expressions of p21, Bax, and PUMA[15–17]; p53 can bind directly to the promoter regions of p21, PUMA, and Bax and induce their transcription[18,19]. In the present study, we found that p21, Bax, and PUMA expressions were markedly induced after SBHA treatment. However, p21, Bax, and PUMA expressions decreased when p53 was inhibited by p53 siRNA, and at the same time, SBHA-induced apoptosis was blocked. All these results indicated that SBHA-induced apoptosis in MCF-7 cells was mediated by the p53 pathway.
In conclusion, we evaluated the contribution of p53 to SBHA-induced apoptosis in MCF-7 cells. We found that SBHA could activate p53 and upregulate its target genes PUMA, p21, and Bax. We also found that the SBHA-induced activation of p53, p21, Bax, and PUMA, as well as apoptosis, was markedly blocked by p53 siRNA. To our knowledge, this is the first study to report that the activation of p53, p21, Bax, and PUMA contributes to the antitumor effects of SBHA. All of these observations suggest that the p53 signaling pathway plays important roles during SBHA-induced apoptosis in MCF-7 cells.
References
- Brinkmann H, Dahler AL, Popa C, Serewko MM, Parsons PG, Gabrielli BG, et al. Histone hyperacetylation induced by histone deacetylase inhibitors not sufficient to cause growth inhibition in human dermal fibroblasts. J Biol Chem 2001;276:22491-9.
- Skarpidi E, Cao H, Heltweg B, White BF, Marhenke RL, Jung M, et al. Hydroxamide derivatives of short-chain fatty acids are potent inducers of human fetal globin gene expression. Exp Hematol 2003;31:197-203.
- Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther 2004;3:425-35.
- Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem Pharmacol 2003;66:1537-45.
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, et al. Features of apoptotic cells measured by flow cytometry. Cytometry 1992;13:795-801.
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239-57.
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000;103:839-42.
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000;6:513-9.
- Moll UM, Zaika A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett 2001;493:65-9.
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004;24:6728-41.
- Ciciarello M, Mangiacasale R, Casenghi M, Limongi MZ, D'Angelo M, Soddu S, et al. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem 2001;276:19205-13.
- Xu ZM, Wang YQ, Mei Q, Chen J, Du J, Wei Y, et al. Effects of p21 (Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998;18:629-43.
- Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, et al. P53-mediated cell cycle arrest and apoptosis through a caspase-3-independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin 2007;28:1057-66.
- Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta 2004; 1705: 3–15.
- Li JH, Li P, Kamut H, Liu FF, Sinicrope FA. Cytotoxic effects of Ad5CMV-P53 expression in two human nasopharyngeal carcinoma cell lines. Clin Cancer Res 1997;3:507-14.
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683-94.
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci 2003;116:4077-85.
- Kaeser MD, Iggo RD. Promoter-specific p53-dependent histone acetylation following DNA damage. Oncogene 2004;23:4007-13.
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell 1995;80:293-9.