
HIV entry inhibitors: a new generation of antiretroviral drugs
Introduction
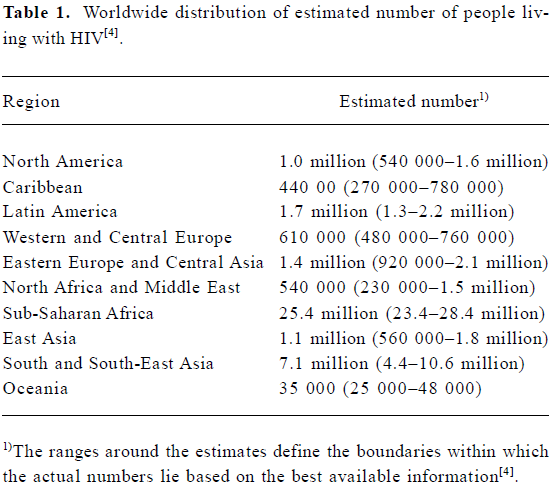
Acquired immunodeficiency syndrome (AIDS) was recognized in 1981, and the first human immunodeficiency virus (HIV) was isolated 2 years later, heralding a new era in the fight against pathogenic viruses[1,2]. Since then, HIV infection has become a major public health problem worldwide, with an estimated 39.4 million infected people as at the end of 2004 (Table 1)[3]. According to the Joint United Nations Programme on HIV/AIDS (UNAIDS) epidemic update, in 2004 there were more than 3.1 million AIDS deaths, including 500 000 children under 15 years of age[4]. The prevalence of HIV-1 is greater in developing countries, and especially in Sub-Saharan Africa, where the infrastructure to prevent and treat the infection is limited[5]. These “hotspots” absorb most of the attention of international committees and organizations, and a significant part of the funding for AIDS prevention and treatment goes towards attempting to scale up antiretroviral (ARV) therapy in developing and transitional countries[6].
HIV is a lentinovirus that is predominantly transmitted by sexual contact, as virus particles can cross the mucosal epithelium and infect specific cells[7,8] expressing the CD4 receptor. Cells bearing CD4 receptors on their membrane belong to the macrophage/monocyte lineage and to a subset of T-cells[9,10]. Initial indications were that HIV-1 used only CD4 to identify and enter the target cells. Soon, however, it became apparent that additional co-receptors were probably required in order for the virus to complete cell entry. Subsequently, several such potential co-receptors were proposed[11,12], but the CCR5 and CXCR4 chemokine receptors are today considered to be the major co-receptors for HIV-1 entry[13–15]. T-cell tropic HIV strains use mainly CXCR4 as a co-receptor and are called X4 strains, whereas macrophage-tropic strains, responsible for host-to-host transmission, use CCR5 as a co-receptor, and are referred to as R5 strains. Thus, macrophages are the principal targets for the establishment of the infection in new individuals[16]. Although it is not always the case[17,18], the transition from viral isolates that use the CCR5 receptor to isolates that use the CXCR4 receptor has been linked with the transition from the latent asymptomatic phase to the clinical manifestations associated with AIDS[19,20].
The most striking feature of HIV-1 infection is the gradual depletion of circulating CD4+ T cells, which leads to increased sensitivity of the patient to opportunistic and chronic infections and to oncogenesis. The cause of the CD4+ T cell depletion is still under debate[21–23]. It is generally accepted, however, that during the asymptomatic phase the daily replenishment rate of CD4+ T cells is much higher than the turnover of infective virus particles for the cytopathicity model to explain the progressive depletion of CD4+ T cells from circulation[24]. An alternative hypothesis proposes that certain viral components contribute to dysfunction of a vital immune mechanism[25].
Over the past 23 years, the main objective in the field of HIV research has been the discovery of drugs that will combat the disease. Satisfactory progress has already been made and there are now more than 20 anti-HIV drugs approved by the American Food and Drug Administration (FDA)[26]. ARV drugs are categorized according to their mode of action into three main groups: 1) the nucleoside reverse transcriptase inhibitors (NRTI); 2) the non-nucleoside reverse transcriptase inhibitors (NNRTI)[27,28]; and 3) the protease inhibitors (PI)[29]. ARV drugs from these categories are now administered in combination (as cocktails) to produce more efficient treatment[30]. This type of therapy, termed “highly active antire-troviral therapy” or HAART, has markedly decreased morta-lity and morbidity in the developed world. Efforts have been made by the World Health Organization (WHO) and UNAIDS to substantially increase the number of people on HAART in developing and transitional countries[6].
Despite the fact that current antiviral treatments have improved prognosis, drug resistance and high toxicity are serious limitations to current treatments that justify the continuation of research efforts for new strategies and interventions[31,32]. Today, AIDS is treatable, and patients can have a good prognosis, but it is still not curable. A new generation of drugs was recently introduced that inhibit viral cell entry (to be discussed later). HIV entry inhibitors appear to be a rational step forward in ARV therapy, because they prevent the virus from infecting new host cells, and may potentially stop or significantly limit HIV transmission[33–35]. In order to rationally design effective drugs, the pathophysiology of HIV must be better understood for ARV therapy research to target specific events in the biology of the virus within the host cell[36,37].
HIV entry
HIV-1 predominately infects cells that have the CD4 receptor on their surface membrane, although this is not always the case[38,39]. Achievement of infection of these cells involves three discrete steps: viral attachment, then co-receptor binding, and finally fusion (see Figure 1). Recognition of the “correct” target cell and attachment to it is primarily achieved through envelope glycoprotein gp120, which binds to CD4 molecules. Gp120 is generated within the infected host cell after cleavage of gp160 by cellular proteases into two functional proteins: gp120 and gp41. It consists of 5 variable (V1-V5) and 5 conserved (C1-C5) regions[40]. Gp120 and gp41 are glycosylated in the Golgi apparatus, and then transported to the membrane that is later incorporated in the viral envelope during the budding of the viral particles to form mature viruses[41]. The envelope membrane is studded with trimers of gp120-gp41 heterodimers, where gp41 forms the cytosolic part and gp120 the extracellular part[42].
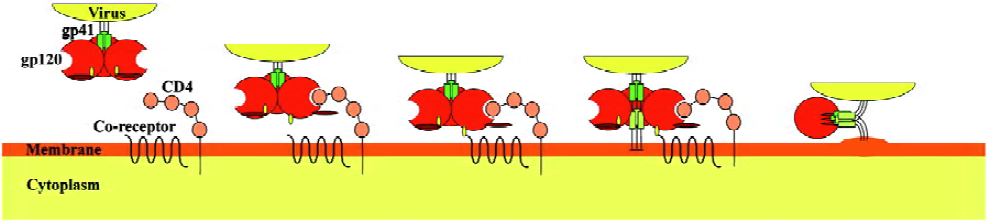
Binding of viral gp120 to host cell CD4 is achieved through interactions of several conserved gp120 residues with the second complementarity-determining region (CDR2) of CD4[43,44]. This interaction alone is not sufficient to achieve cell entry, but it is necessary in order to identify the target cell and also to increase the affinity of other viral components for the co-receptor molecules. Indeed, binding of gp120 to CD4 causes conformational changes to the variable loop regions V1/V2 and V3 of gp120, causing the V3 loop to evaginate, thus becoming exposed to the co-receptors[45] (Figure 1). The major co-receptors that HIV-1 uses are the CCR5 and CXCR4 chemokine receptors. The exact mechanism of interaction between the variable loop regions V1/V2 and V3 and the chemokine receptors is not well understood and it merits a more detailed investigation. It has been suggested, however, that the interaction between V3 and CCR5 is ionic in nature, and results in enhancement of the process of activation-induced cell death of responding effector CD4+ T cells during antigen presentation[22,46,47].
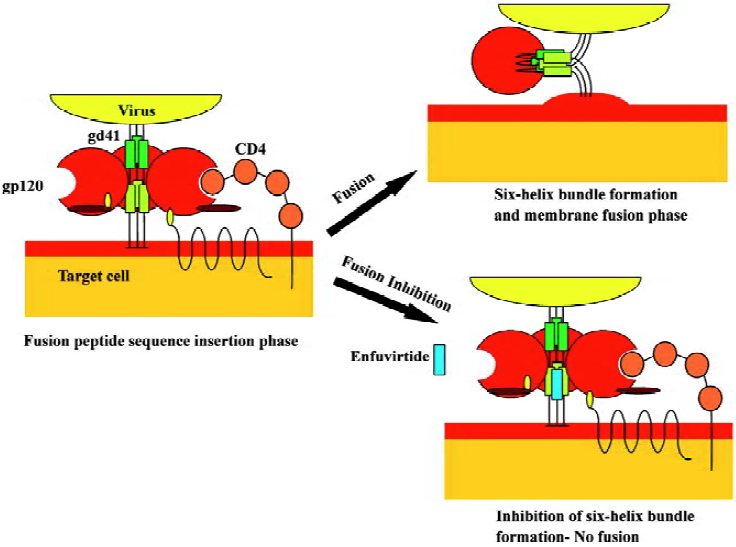
The final step for viral entry requires fusion of the viral envelope components with the target surface membrane; this is achieved with the use of gp41, which is a glycoprotein consisting of 3 main domains: an intracellular domain (endodomain), a transmembrane anchor and an extracellular domain (ectodomain). The ectodomain is the key structure responsible for fusion and consists of a hydrophobic fusion peptide sequence at the N-terminal, two hydrophobic heptad repeats (HR1 and HR2) at the C-terminal, and a hinge region, where a disulfide-bond loop is formed between the two heptad repeats during fusion[48,49]. On binding of gp120 to CD4 and subsequently to the co-receptor, further conformational changes occur that lead to gp41 dissociation from gp120. The gp41 unfolds and the hydrophobic fusion peptide sequence extends out of the viral membrane towards the host cell membrane. Insertion of the fusion peptide into the host cell membrane leads gp41 to fold into a hairpin-like structure where the two hydrophobic heptad repeats (HR1 and HR2) lie antiparallel, forming a 6-helix bundle[50,51]. This hairpin structure is believed to be responsible for the fusion of the HIV envelope to the host cell membrane.
Enfuvirtide: the first FDA-approved fusion inhibitor
Enfuvirtide (formerly known as T-20) is the first fusion inhibitor approved by the FDA and the European Commission for the Treatment of AIDS, and is available under the trade name Fuzeon (Trimeris and Roche). It is a 36 amino acid synthetic peptide homologous to the HR2 region of gp41 (residues 127-162) [52,53], that has the ability to interfere with the fusion pathway by mimicking the HR2 domain[54]. The accepted mode of action proposes that enfuvirtide targets conformational changes during fusion by binding to the HR1 domain. Recent evidence indicates that enfuvirtide interacts with multiple sites in gp41 and gp120[55]. This binding prevents the formation of the 6-helix bundle by preventing HR2 from refolding antiparallel to HR1[56,57]. Thus, inhibition of fusion of the viral envelope to cell membranes is achieved by blocking a critical step in the fusion pathway (Figure 2).
In the initial stages of discovery, enfuvirtide appeared to inhibit HIV-1 replication very effectively in various cell types and clinical trials proved to be very promising. Phase I/II trials provided proof that HIV entry was inhibited after treating patients with 100 mg enfuvirtide twice daily for 14 d. The levels of plasma HIV RNA after 14 d of treatment demonstrated a 1.96 lg median decline[58]. Phase II clinical trials were performed on 71 HIV-infected individuals who were treated with 50 mg enfuvirtide together with other ARV drugs for 48 weeks. There was a 1.0 log10 decline from baseline in HIV RNA and a median gain of CD4 cell counts of 84.9 cells/μL, with no significant toxicity[59].
Furthermore, two TORO (T-20 vs Optimized Regimens Only) Phase III clinical trials were performed in America (TORO 1) and in Europe and Australia (TORO 2). The trials had similar protocols: they compared the efficacy and safety of enfuvirtide plus an optimized antiretroviral regimen with the efficacy and safety of an optimized antiretroviral regimen alone[60,61]. In both studies the least-squares mean change from baseline in the plasma viral load indicated a significant difference in the decrease in the enfuvirtide group compared with the control (P<0.01). In the same way, the mean count of CD4 cells/mL was significantly greater in the enfuvirtide group compared with the controls (P<0.01).
Further studies are currently being performed on the exact metabolic pathway of enfuvirtide, potential drug resistance problems, and identification of synergistic interactions with other drugs. Several reports concluded that enfuvirtide does not appear to interfere with the activities of cytochrome P450, probably because it is a peptide and is easily hydrolyzed in the body[62,63]. Enfuvirtide was found to act synergistically with other potential entry inhibitors in vitro, such as AMD3100 and PRO542, producing results that encouraged the use of combinations of entry inhibitors as part of a new generation of ARV strategies[64,65]. However, HIV resistance has been reported in patients treated with enfuvirtide, indicating a hotspot from codons 36 to 38 of the HR1 domain[66], as well as other sites in gp41[67–69]. Additionally, primary resistance has been reported, which appears to be more frequent than predicted[70], indicating that more research is needed in this field.
Enfuvirtide obtained accelerated approval by the FDA in 2003 and became the 17th licensed ARV drug and the first to inhibit HIV entry. The drug is supplied as a lyophilized powder in single-dose vials containing 108 mg of the drug. Reconstitution of the powder in 1.1 mL sterile water for injection produces a single dose of 90 mg/mL[71] that is injected subcutaneously. Enfuvirtide has two currently known major drawbacks. First, being a peptide, it can only be administered by injection and not orally. This makes usage more difficult, because patients must be educated for self-administration. Second, the cost of enfuvirtide is high, because it is a synthetic peptide that is manufactured by a highly complicated process involving large amounts of raw materials[72,73]. It is estimated that the annual cost of enfuvirtide therapy is approximately US$20 000 per patient, and if taken in combination with other ARV drugs then the cost of therapy could approach US$30 000.
Potential drugs targeting entry and fusion
Attachment inhibitors Current novel antiretroviral drugs aim to interfere with the crucial HIV entry steps: viral attachment, co-receptor binding and fusion. One approach for interfering with viral attachment involves the use of a tetravalent fusion protein construct, consisting of a human IgG2 in which the Fv portions of both the heavy and light chains have been replaced with the D1 and D2 domains of human CD4[74,75]. This CD4-immunoglobulin fusion construct, called PRO 542, is suggested to bind to the viral gp120 and thus prevent the virus from interacting with CD4-bearing host cells. Phase I clinical trials indicated that PRO 542 has a half-life of 3–4 d when a relatively high dose was used (10 mg/kg), and no dose-limiting toxicities were observed[76]. In addition, in phase II clinical trials, 12 HIV-infected patients were treated with 25 mg/kg single-dose PRO 542. The drug was well tolerated and the acute reduction caused in the HIV-1 RNA was statistically significant, even in patients with advanced AIDS[77].
In the same way, several other compounds target either the gp120 or the CD4 receptor and interfere with HIV attachment. FP-21399 is a bis(disulfonapthelene) derivative that binds to gp120, most probably near the third variable domain, because interactions with antibodies against the V3 loop region were blocked[78]. A phase I study showed that it caused an increase in CD4 cell counts, and a significant decrease in viral load and minor side effects[79]. BMS-378806, a 4-methoxy-7-azaindole derivative, is a compound that can be administered orally, and was developed by Bristol Myers Squibb[80,81]. Despite the fact that phase I and II studies showed promising results, Bristol Myers Squibb decided to investigate similar drugs such as BMS-488043, an analogue of BMS-378806, in order to optimize its effectiveness[82]. A series of polyanionic compounds, for example dextrin-2-sulfate, Carraguard and PRO 2000 are in clinical trials, and are designed to be topically administered[83–85]. Finally, TNX-355, a humanized anti-CD4 mAB that binds to CD4 without interfering with its biological function, significantly decreased viral load and increased CD4 cell counts in a phase I trial[86].
Co-receptor binding inhibitors The most interesting target in HIV entry is the co-receptor binding phase. Current drug research is focused on designing compounds that prevent the virus interacting with the chemokine receptors. The CCR5 receptor is the principal target, and a number of potential drugs are currently being studied. SCH-C is a small mo-lecule that inhibits the binding of gp120 to CCR5, and initial in vitro experiments have indicated good inhibitory activity against R5 viruses as well as synergistic effects with several ARV drugs, including enfuvirtide[87,88]. Although it can be administered orally and clinical studies showed decreased viral loads, electrocardiographic anomalies due to arrhy-thmias were reported at high dosages[89]. Another compound, SCH-D, has been found to have greater in vitro and in vivo antiviral properties, with no apparent side effects. Clinical studies for this drug are still ongoing[90]. Interestingly, it was recently reported that V3-like peptides from X4 strains with more electropositive V3 domains were effective antagonists and potential infectivity blockers of R5 variants[91].
TAK-779 was the first non-peptidic molecule found to inhibit co-receptor attachment by binding to CCR5 at transmembrane helices 1, 2, 3, and 7[92,93]. It has the disadvantage of intravenous administration and because of irritations observed at the injection site, its development was discontinued. It was replaced by another compound, T-220, which can be administered orally, and shows promising anti-R5 HIV acti-vity[94]. Similarly, UK-427,857 is a novel CCR5 inhibitor that has acceptable pharmacokinetic and metabolic rates in mice, rats, dogs and humans, and can be administered orally[95]. Finally, PRO 140 is one of the few monoclonal antibodies that has been used as an entry inhibitor and has been reported to block co-receptor attachment without down-modulating or inducing signaling of the CCR5 chemokine receptor[96,97].
CXCR4, the second major HIV co-receptor, is also a target for current drug research. AMD-3100, one of the first entry inhibitors, was found to inhibit viral entry well before the discovery of co-receptor usage by HIV[98]. It is a bicyclam compound of low molecular weight that inhibits the electrostatic interaction between CXCR4 and gp120 by ionic binding to the second extracellular loop (ECL2) and the adjacent membrane-spanning domain (TM4) of the CXCR4 receptor[99]. Despite the fact that in phase I and II clinical trials, intravenous administration of AMD-3100 significantly reduced the viral load[100], it was later replaced by an orally available compound, AMD-070. A non-peptidic compound, KRH-1636, which is absorbed through the duodenum, had similar efficacies to AMD-3100[101]. Finally, T-22 and ALX40-4C are positively charged peptides that occupy the V3 region and competitively inhibit binding of gp120 to the negatively charged amino acid residues on CXCR4[102–104].
In conclusion, the role of the V3 region in the mechanism of cell attachment and entry in relation to the major co-receptors is being actively pursued. In addition to biological studies, physicochemical studies on the interacting protein domains are being carried out in an attempt to decipher the interface conformations between the virus and the cell[105].
Fusion inhibitors Understanding the mechanism of fusion of the viral envelope with the host membrane played a crucial role in the development of new generation ARV drugs. This became apparent when enfuvirtide was licensed as the first viral entry inhibitor, and it is currently used in HAART. Resistance to enfuvirtide has been reported, which has led to the design of a second generation HR2 mimetic peptide. T-1249 is a 39-L-amino acid synthetic peptide that contains a pocket-binding sequence that makes the HR1 and HR2 interaction more stable. Studies on T-1249 showed that it has greater efficacy and longer half-life than enfuvirtide. Additionally, efficacy against enfuvirtide-resistant viruses has been reported, indicating that this second generation fusion inhibitor is a step forward[106,107]. However, Roche and Trimeris decided to halt clinical development due to formulation concerns[108].
5-Helix is a newly designed recombinant C-peptide that consists of 5 of the 6 helices that are formed during the fusion phase. A CHR domain is missing for the 6-helix bundle formation, and thus there is one exposed groove. This groove binds to a CHR domain in gp41 and inhibits fusion of the viral membrane to the host membrane[109]. Because it is a recombinant peptide, it has a much lower cost of production compared with the synthetic enfuvirtide. Initial studies demonstrated potent antiretroviral activity, with IC50 values in the low nanomolar range[110].
Finally, N-peptides represent another group of peptides with potential inhibitory effects against HIV entry. Initial studies have indicated that they are weaker inhibitors than the C-peptides, with IC50 values in the micromolar range. However, chimeric molecules composed of soluble trimeric coiled coils have shown promising results. IQN17 is one of the first such peptides with potent inhibitory effects, and the current most potent chimeric N-peptide, IQN23, is reported to have an IC50 value of 15 nmol/L[111].
Conclusion
Antiretroviral chemotherapy has recently acquired a new “weapon” in the fight against AIDS. Enfuvirtide is the first HIV entry inhibitor that was approved by FDA, and it is currently used in combination with other ARV drugs. Results from clinical trials indicated that it had potent activity against HIV strains that are resistant to other ARV drugs, although some resistance to enfuvirtide has been reported. The design of other entry inhibitors has moved forward, and every phase of HIV entry is actively pursued as a target for potential inhibitors. Probably the most exciting prospect is potential interference with co-receptor usage, particularly that of CCR5.
ARV drug development aims to produce drugs with potent antiretroviral activity, with IC50 values in the nanomolar range, with no or limited toxicity and that can be administered orally. Several compounds are currently in clinical trials, and we are optimistic that new, more effective drugs will be added to the ARV armory.
References
- Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983;220:868-71.
- Gallo RC, Sarin PS, Gelmann EP, Robert-Guroff M, Richardson E, Kalyanaraman VS, et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983;220:865-7.
- HIV/AIDS Facts and figures [database on the internet]. New Delhi: WHO Reginal Office for South-East Asia . [cited 2005 Apr 20]. Available from: http://w3.whosea.org/EN/Section10/Section18/Section348.htm#Global
- UNAIDS/WHO. AIDS Epidemic Update. Geneva: UNAIDS; 2004.
- Gayle HD, Hill GL. Global impact of human immunodeficiency virus and AIDS. Clin Microbiol Rev 2001;14:327-35.
- UNAIDS/WHO. ‘3 by 5’ Progress Report. France: WHO; 2005.
- Bomsel M. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat Med 1997;3:42-7.
- Ullrich R, Schmidt W, Zippel T, Schneider T, Zeitz M, Riecken EO. Mucosal HIV infection. Pathobiology 1998;66:145-50.
- Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984;312:763-7.
- Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, et al. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 1984;312:767-8.
- Rucker J, Samson M, Doranz BJ, Libert F, Berson JF, Yi Y, et al. Regions in beta-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell 1996;87:437-46.
- Roderiquez G, Oravecz T, Yanagishita M, Bou-Habib DC, Mostowski H, Norcross MA. Mediation of human immunodeficiency virus type 1 binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120-gp41. J Virol 1995;69:2233-9.
- Berson JF, Long D, Doranz BJ, Rucker J, Jirik FR, Doms RW. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strains. J Virol 1996;70:6288-95.
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996;381:661-6.
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996;381:667-73.
- van’t Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, Scherpbier HJ, Veenstra J, et al. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J Clin Invest 1994;94:2060-7.
- de Roda Husman AM, van Rij RP, Blaak H, Broersen S, Schuitemaker H. Adaptation to promiscuous usage of chemokine receptors is not a prerequisite for human immunodeficiency virus type 1 disease progression. J Infect Dis 1999;180:1106-15.
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, et al. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol 1992;66:1354-60.
- Bjorndal A, Sonnerborg A, Tscherning C, Albert J, Fenyo EM. Phenotypic characteristics of human immunodeficiency virus type 1 subtype C isolates of Ethiopian AIDS patients. AIDS Res Hum Retroviruses 1999;15:647-53.
- Richman DD, Bozzette SA. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J Infect Dis 1994;169:968-74.
- Roumier T, Castedo M, Perfettini JL, Andreau K, Metivier D, Zamzami N, et al. Mitochondrion-dependent caspase activation by the HIV-1 envelope. Biochem Pharmacol 2003;66:1321-9.
- Zafiropoulos A, Baritaki S, Sioumpara M, Spandidos DA, Krambovitis E. V3 induces in human normal cell populations an accelerated macrophage-mediated proliferation: apoptosis phenomenon of effector T cells when they respond to their cognate antigen. Biochem Biophys Res Commun 2001;281:63-70.
- Dockrell DH, Badley AD, Algeciras-Schimnich A, Simpson M, Schut R, Lynch DH, et al. Activation-induced CD4+ T cell death in HIV-positive individuals correlates with Fas susceptibility, CD4+ T cell count, and HIV plasma viral copy number. AIDS Res Hum Retroviruses 1999;15:1509-18.
- Graziosi C, Pantaleo G, Butini L, Demarest JF, Saag MS, Shaw GM, et al. Kinetics of human immunodeficiency virus type 1 (HIV-1) DNA and RNA synthesis during primary HIV-1 infection. Proc Natl Acad Sci USA 1993;90:6405-9.
- Krambovitis E, Zafiropoulos A, Baritaki S, Spandidos DA. Simple electrostatic interaction mechanisms in the service of HIV-1 pathogenesis. Scand J Immunol 2004;59:231-4.
- De Clercq E. Antiviral drugs in current clinical use. J Clin Virol 2004;30:115-33.
- Imamichi T. Action of anti-HIV drugs and resistance: reverse transcriptase inhibitors and protease inhibitors. Curr Pharm Des 2004;10:4039-53.
- Zapor MJ, Cozza KL, Wynn GH, Wortmann GW, Armstrong SC. Antiretrovirals, Part II: focus on non-protease inhibitor antiretrovirals (NRTIs, NNRTIs, and fusion inhibitors). Psychosomatics 2004;45:524-35.
- Dunn BM, Goodenow MM, Gustchina A, Wlodawer A. Retroviral proteases. Genome Biol 2002; 3: REVIEWS 3006.
- Pereira CF, Paridaen JT. Anti-HIV drug development: an overview. Curr Pharm Des 2004;10:4005-37.
- Johnson VA, Brun-Vezinet F, Clotet B, Conway B, D’Aquila RT, Demeter LM, et al. Update of the drug resistance mutations in HIV-1: 2004. Top HIV Med 2004;12:119-24.
- van Heeswijk RP. Optimized antiretroviral therapy: the role of therapeutic drug monitoring and pharmacogenomics. Expert Rev Anti Infect Ther 2003;1:75-81.
- Stone A. Microbicides: a new approach to preventing HIV and other sexually transmitted infections. Nat Rev Drug Discov 2002;1:977-85.
- Moore JP, Shattock RJ. Preventing HIV-1 sexual transmission: not sexy enough science, or no benefit to the bottom line? J Antimicrob Chemother 2003;52:890-2.
- Shattock RJ, Moore JP. Inhibiting sexual transmission of HIV-1 infection. Nat Rev Microbiol 2003;1:25-34.
- Sleasman JW, Goodenow MM. 13. HIV-1 infection. J Allergy Clin Immunol 2003;111:S582-92.
- De Clercq E. HIV-chemotherapy and -prophylaxis: new drugs, leads and approaches. Int J Biochem Cell Biol 2004;36:1800-2.
- Zerhouni B, Nelson JA, Saha K. CXCR4-dependent infection of CD8+, but not CD4+, lymphocytes by a primary human immunodeficiency virus type 1 isolate. J Virol 2004;78:12288-96.
- Zerhouni B, Nelson JA, Saha K. Isolation of CD4-independent primary human immunodeficiency virus type 1 isolates that are syncytium inducing and acutely cytopathic for CD8+ lymphocytes. J Virol 2004;78:1243-55.
- Farzan M, Choe H, Desjardins E, Sun Y, Kuhn J, Cao J, et al. Stabilization of human immunodeficiency virus type 1 envelope glycoprotein trimers by disulfide bonds introduced into the gp41 glycoprotein ectodomain. J Virol 1998;72:7620-5.
- Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem 1990;265:10373-82.
- Yang X, Farzan M, Wyatt R, Sodroski J. Characterization of stable, soluble trimers containing complete ectodomains of human immunodeficiency virus type 1 envelope glycoproteins. J Virol 2000;74:5716-25.
- Olshevsky U, Helseth E, Furman C, Li J, Haseltine W, Sodroski J. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J Virol 1990;64:5701-7.
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998;393:648-59.
- Sullivan N, Sun Y, Sattentau Q, Thali M, Wu D, Denisova G, et al. CD4-Induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J Virol 1998;72:4694-703.
- Zafiropoulos A, Baritaki S, Vlata Z, Spandidos DA, Krambovitis E. Dys-regulation of effector CD4+ T cell function by the V3 domain of the HIV-1 gp120 during antigen presentation. Biochem Biophys Res Commun 2001;284:875-9.
- Baritaki S, Zafiropoulos A, Sioumpara M, Politis M, Spandidos DA, Krambovitis E. Ionic interaction of the HIV-1 V3 domain with CCR5 and deregulation of T lymphocyte function. Biochem Biophys Res Commun 2002;298:574-80.
- Bernstein HB, Tucker SP, Kar SR, McPherson SA, McPherson DT, Dubay JW, et al. Oligomerization of the hydrophobic heptad repeat of gp41. J Virol 1995;69:2745-50.
- Chambers P, Pringle CR, Easton AJ. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J Gen Virol 1990;71:3075-80.
- Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol 1995;2:1075-82.
- Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997;387:426-30.
- Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci USA 1992;89:10537-41.
- Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA 1994;91:9770-4.
- Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J Virol 1995;69:3771-7.
- Liu S, Lu H, Niu J, Xu Y, Wu S, Jiang S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem 2005;280:11259-73.
- Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR Jr, Merutka G. HIV-1 membrane fusion mechanism: structural studies of the interactions between biologically-active peptides from gp41. Biochemistry 1996;35:13697-708.
- Furuta RA, Wild CT, Weng Y, Weiss CD. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol 1998;5:276-9.
- Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 1998;4:1302-7.
- Lalezari JP, Eron JJ, Carlson M, Cohen C, DeJesus E, Arduino RC, et al. A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. Aids 2003;17:691-8.
- Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 2003;348:2175-85.
- Lazzarin A, Clotet B, Cooper D, Reynes J, Arasteh K, Nelson M, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med 2003;348:2186-95.
- Zhang X, Lalezari JP, Badley AD, Dorr A, Kolis SJ, Kinchelow T, et al. Assessment of drug-drug interaction potential of enfuvirtide in human immunodeficiency virus type 1-infected patients. Clin Pharmacol Ther 2004;75:558-68.
- Ruxrungtham K, Boyd M, Bellibas SE, Zhang X, Dorr A, Kolis S, et al. Lack of interaction between enfuvirtide and ritonavir or ritonavir-boosted saquinavir in HIV-1-infected patients. J Clin Pharmacol 2004;44:793-803.
- Tremblay CL, Kollmann C, Giguel F, Chou TC, Hirsch MS. Strong in vitro synergy between the fusion inhibitor T-20 and the CXCR4 blocker AMD-3100. J Acquir Immune Defic Syndr 2000;25:99-102.
- Nagashima KA, Thompson DA, Rosenfield SI, Maddon PJ, Dragic T, Olson WC. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J Infect Dis 2001;183:1121-5.
- Rimsky LT, Shugars DC, Matthews TJ. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 1998;72:986-93.
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 2002;46:1896-905.
- Poveda E, Rodes B, Toro C, Martin-Carbonero L, Gonzalez-Lahoz J, Soriano V. Evolution of the gp41 env region in HIV-infected patients receiving T-20, a fusion inhibitor. Aids 2002;16:1959-61.
- Xu L, Pozniak A, Wildfire A, Stanfield-Oakley SA, Mosier SM, Ratcliffe D, et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob Agents Chemother 2005;49:1113-9.
- Carmona R, Perez-Alvarez L, Munoz M, Casado G, Delgado E, Sierra M, et al. Natural resistance-associated mutations to Enfuvirtide (T20) and polymorphisms in the gp41 region of different HIV-1 genetic forms from T20 naive patients. J Clin Virol 2005;32:248-53.
- Fuzeon [package insert]. Injection Instructions. Durham, NC, and Nutley, NJ: Trimeris Inc and Roche Laboratories Inc; 2003.
- Steinbrook R. HIV infection: a new drug and new costs. N Engl J Med 2003;348:2171-2.
- Tashima KT, Carpenter CC. Fusion inhibition: a major but costly step forward in the treatment of HIV-1. N Engl J Med 2003;348:2249-50.
- Allaway GP, Davis-Bruno KL, Beaudry GA, Garcia EB, Wong EL, Ryder AM, et al. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res Hum Retroviruses 1995;11:533-9.
- Trkola A, Pomales AB, Yuan H, Korber B, Maddon PJ, Allaway GP, et al. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J Virol 1995;69:6609-17.
- Jacobson JM, Lowy I, Fletcher CV, O’Neill TJ, Tran DN, Ketas TJ, Trkola A, et al. Single-dose safety, pharmacology, and antiviral activity of the human immunodeficiency virus (HIV) type 1 entry inhibitor PRO 542 in HIV-infected adults. J Infect Dis 2000;182:326-9.
- Jacobson JM, Israel RJ, Lowy I, Ostrow NA, Vassilatos LS, Barish M, et al. Treatment of advanced human immunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542. Antimicrob Agents Chemother 2004;48:423-9.
- Ono M, Wada Y, Wu Y, Nemori R, Jinbo Y, Wang H, et al. FP-21399 blocks HIV envelope protein-mediated membrane fusion and concentrates in lymph nodes. Nat Biotechnol 1997;15:343-8.
- Dezube BJ, Dahl TA, Wong TK, Chapman B, Ono M, Yamaguchi N, et al. A fusion inhibitor (FP-21399) for the treatment of human immunodeficiency virus infection: a phase I study. J Infect Dis 2000;182:607-10.
- Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci USA 2003;100:11013-8.
- Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci USA 2004;101:5036-41.
- Hanna G, Lalezari J, Hellinger J, Wohl D, Masterson T, Fiske W, . Antiviral activity, safety, and tolerability of a novel, oral small-molecule HIV-1 attachment inhibitor, BMS-488043, in HIV-1-infected subjects. In: Proceedings of the 11th conference on retroviruses and opportunistic infections; 2004 Feb 8-11, San Francisco. 2004.
- Shaunak S, Thornton M, John S, Teo I, Peers E, Mason P, et al. Reduction of the viral load of HIV-1 after the intraperitoneal administration of dextrin 2-sulphate in patients with AIDS. Aids 1998;12:399-409.
- Dezzutti CS, James VN, Ramos A, Sullivan ST, Siddig A, Bush TJ, et al. In vitro comparison of topical microbicides for prevention of human immunodeficiency virus type 1 transmission. Antimicrob Agents Chemother 2004;48:3834-44.
- Morrow K, Rosen R, Richter L, Emans A, Forbes A, Day J, et al. The acceptability of an investigational vaginal microbicide, PRO 2000 Gel, among women in a phase I clinical trial. J Womens Health (Larchmt) 2003;12:655-66.
- Kuritzkes DR, Jacobson J, Powderly WG, Godofsky E, DeJesus E, Haas F, et al. Antiretroviral activity of the anti-CD4 monoclonal antibody TNX-355 in patients infected with HIV type 1. J Infect Dis 2004;189:286-91.
- Tremblay CL, Giguel F, Kollmann C, Guan Y, Chou TC, Baroudy BM, et al. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob Agents Chemother 2002;46:1336-9.
- Tsamis F, Gavrilov S, Kajumo F, Seibert C, Kuhmann S, Ketas T, et al. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J Virol 2003;77:5201-8.
- Reynes J, Rouzier R, Kanouni T, Baillat V, Baroudy B, Keung A. SCH C: Safety and antiviral effects of a CCR5 receptor antagonist in HIV-1-infected subjects. In: Proceedings of the 9th conference on retroviruses and opportunistic infections; 2002 Feb 24-28, San Francisco. 2002
- Schurmann D, Rouzier R, Nougarede R, Reynes J, Fatkenheuer G, Raffi F, . SCH D: Antiviral activity of a CCR5 receptor antagonist. In: Proceedings of the 11th conference on retroviruses and opportunistic infections, 2004 Feb 8-11, San Francisco. 2004.
- Baritaki S, Dittmar MT, Spandidos DA, Krambovitis E. In vitro inhibition of R5 HIV-1 infectivity by X4 V3-derived synthetic peptides. Int J Mol Med 2005;16:333-6.
- Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA 1999;96:5698-703.
- Dragic T, Trkola A, Thompson DA, Cormier EG, Kajumo FA, Maxwell E, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci USA 2000;97:5639-44.
- Iizawa Y, Kanzaki N, Takashima K, Miyake H, Tagawa Y, Sugihara Y, . Anti-HIV-1 Activity of TAK-220, a Small Molecule CCR5 Antagonist. In: Proceedings of the 10th conference on retroviruses and opportunistic infections, 2003 Feb 10-14, San Francisco. 2003.
- Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman AN, Smith DA. Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab Dispos 2005;33:587-95.
- Olson WC, Rabut GE, Nagashima KA, Tran DN, Anselma DJ, Monard SP, et al. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J Virol 1999;73:4145-55.
- Trkola A, Ketas TJ, Nagashima KA, Zhao L, Cilliers T, Morris L, et al. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J Virol 2001;75:579-88.
- de Clercq E, Yamamoto N, Pauwels R, Balzarini J, Witvrouw M, De Vreese K, et al. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob Agents Chemother 1994;38:668-74.
- Labrosse B, Brelot A, Heveker N, Sol N, Schols D, De Clercq E, et al. Determinants for sensitivity of human immunodeficiency virus coreceptor CXCR4 to the bicyclam AMD3100. J Virol 1998;72:6381-8.
- Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother 2000;44:1667-73.
- Ichiyama K, Yokoyama-Kumakura S, Tanaka Y, Tanaka R, Hirose K, et al. A duodenally absorbable CXC chemokine receptor 4 antagonist, KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA 2003;100:4185-90.
- Doranz BJ, Grovit-Ferbas K, Sharron MP, Mao SH, Goetz MB, Daar ES, et al. A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J Exp Med 1997;186:1395-400.
- Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, et al. A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med 1997;186:1389-93.
- Murakami T, Zhang TY, Koyanagi Y, Tanaka Y, Kim J, Suzuki Y, et al. Inhibitory mechanism of the CXCR4 antagonist T22 against human immunodeficiency virus type 1 infection. J Virol 1999;73:7489-96.
- Galanakis PA, Spyroulias GA, Rizos A, Samolis P, Krambovitis E. Conformational properties of HIV-1 gp120/V3 immunogenic domains. Curr Med Chem 2005;12:551-68.
- Eron JJ, Gulick RM, Bartlett JA, Merigan T, Arduino R, Kilby JM, et al. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J Infect Dis 2004;189:1075-83.
- Lalezari JP, Bellos NC, Sathasivam K, Richmond GJ, Cohen CJ, Myers RA, et al. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J Infect Dis 2005;191:1155-63.
- Martin-Carbonero L. Discontinuation of the clinical development of fusion inhibitor T-1249. AIDS Rev 2004;6:61-3.
- Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science 2001;291:884-8.
- Root MJ, Hamer DH. Targeting therapeutics to an exposed and conserved binding element of the HIV-1 fusion protein. Proc Natl Acad Sci USA 2003;100:5016-21.
- Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci USA 2001;98:11187-92.