
Modulation of major voltage- and ligand-gated ion channels in cultured neurons of the rat inferior colliculus by lidocaine1
Introduction
Tinnitus is an auditory phantom sensation without external auditory stimulation, and is a prevalent otological problem that affects a large population of the world[1,2]. Currently, there is no effective therapeutic strategy for the treatment of tinnitus, but sometimes local anesthetics are clinically used to relieve its symptoms[3]. Local anesthetics are a broad family of drugs that are composed of 3 components, including a lipophilic aromatic ring, an intermediate ester or amide chain, and terminal amine. They are classified into 2 groups: the amine group and the ester group. Procaine, an ester-type local anesthetic, was the first local anesthetic used for tinnitus-suppressing effects[4]. Lidocaine, an amine-type local anesthetic currently used for anti-arrhythmic therapy, was then introduced into tinnitus treatment and has been reported to ameliorate tinnitus of sufferers when administered intravenously[5,6]. However, the underlying mechanism for the tinnitus-suppressing effects of lidocaine remains obscure.
The understanding of tinnitus generation will help us to understand how lidocaine relieves tinnitus symptoms. Unfortunately, the neural mechanisms of tinnitus are also poorly understood. Several hypotheses have been proposed for explaining the generation of tinnitus. One of these hypotheses claims that the central auditory system is involved in the perception of tinnitus[7,8]; however, whether the tinnitus generator is located peripherally or centrally is still unknown. More and more researchers tend to believe that, at least in some cases, tinnitus has a central rather than peripheral origin[9–12]. For example, tinnitus still persists in some patients following acoustic removal with sectioning of the auditory nerve[13]. Because the common dose of lidocaine, which can pass the blood–brain barrier immediately after intravenous injection[8,14], is approximately 1.5 mg/kg body weight for tinnitus treatment[3,14-16], a possibility exists that the pharmacological target of lidocaine therapy for tinnitus with a central origin is located in the central auditory system. Some studies suggest direct pharmacological actions of lidocaine on the central auditory system[17]. For example, the V wave of the auditory brainstem response[18], which is associated with the activity of the inferior colliculus (IC), and salicylate-induced discharge of IC neurons[19] can be inhibited by lidocaine. There is also a suppressive effect of lidocaine on tinnitus in patients following the translabyrinthine removal of vestibular schwannoma[3], suggesting a central target of lidocaine. The purpose of the present study was to explore how lidocaine, as a therapeutic drug for tinnitus, targets ion channels and changes the excitability of central auditory neurons. Because evidence from a number of studies suggests that the IC is a potential candidate for the locus of tinnitus generation[19–21], we performed experiments in primary cultured neurons of rat IC with whole-cell patch-clamp techniques to examine how lidocaine affects the functions of major voltage- and ligand-gated channels, as well as current-evoked neuronal firing in this auditory region.
Materials and methods
Cell culture The use and care of the animals in this study followed the guidelines and protocols of the Institutional Animal Care and Use Committee of University of Science and Technology of China.
Primary cultures of dissociated neonatal rat IC neurons were prepared as described previously[22]. In brief, Wistar rats, aged P0 (either sex), were decapitated; the whole brain was transferred to iced Hanks’ solution. The IC (Figure 1A) was then collected and placed under a microscope. The tissue was incubated with 0.25% trypsin (Sigma, St Louis, MO, USA) for 15 min at 37 °C and mechanically dissociated by trituration with a Pasteur pipette in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA). The isolated neurons were plated (1.5×106 cell/mL) on poly-L-lysine (Sigma, USA)-coated cover glasses and grown in DMEM with L-glutamine plus 10% fetal bovine serum, 10% F-12 nutrient mixture, 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco, USA) for 24 h. Neuron-basal medium (1.5 mL) with 2% B27 (Gibco, USA) was replaced every 3–4 d. Treatment with 5-fluoro-5´-deoxyuridine (20 µg/mL; Sigma, USA) on d 4 after plating was used to block the cell division of non-neuronal cells, which helped to stabilize the cell population. The cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere. Cells were used for electrophysiological recordings 7–20 d after plating (Figure 1B).
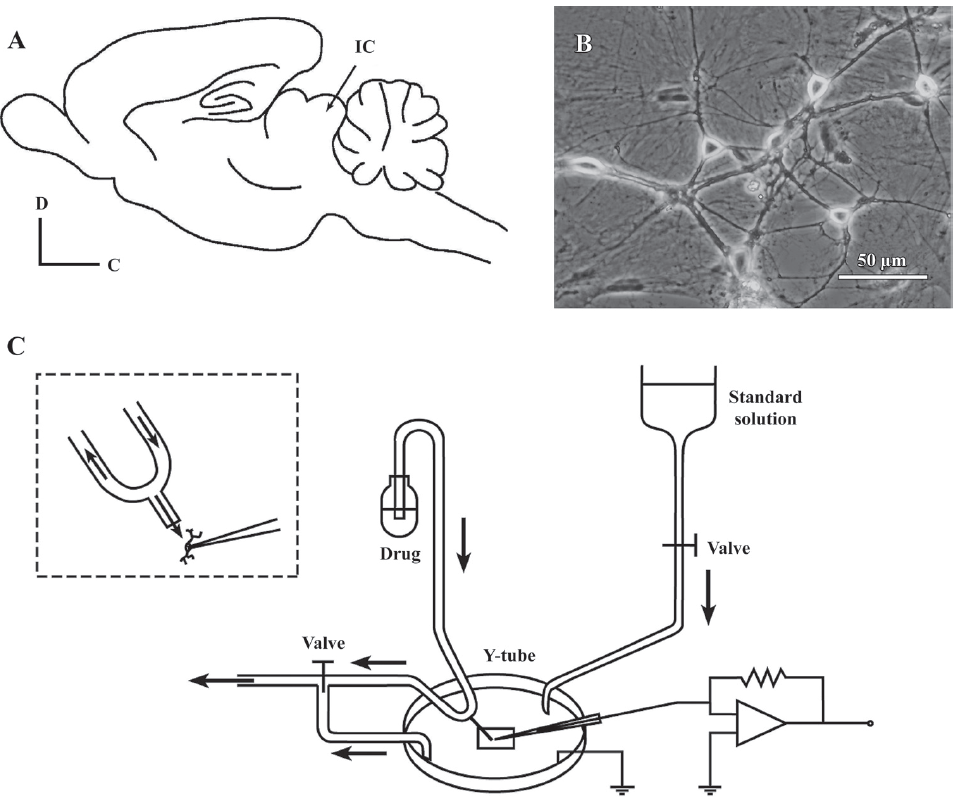
Solutions and drugs The standard external solution contained (in mmol/L): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, and was titrated to a pH of 7.4 with Tris base. The standard external solution was used for recording glycine-induced currents (IGly), gamma aminobutyric acid (GABA)-induced currents (IGABA), and the current-evoked firing of action potentials. The external solution for recording aspartate (Asp)-induced currents (IAsp) contained (in mmol/L): 150 NaCl, 5 KCl, 2 CaCl2, 10 glucose, 10 HEPES, and 1 µmol/L glycine, and was titrated to a pH of 7.4 with Tris base. The external solution for recording Na+ currents (INa) contained (in mmol/L): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, 5 4-aminopyridine (4-AP), 0.2 CdCl2, and 0.1 NiCl2, and was titrated to a pH of 7.4 with Tris base. The external solution for recording transient outward K+ currents (IA) contained (in mmol/L): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, 0.0003 tetrodotoxin (TTX), 0.2 CdCl2, and 20 tetraethylammonium chloride (TEACl), and was titrated to a pH of 7.4 with Tris base. The osmolarity of all the bath solutions was adjusted to 310–320 mOsm/L with sucrose and a microosmometer (model 3300; Advanced Instruments, Pomona, CA, USA).
The patch pipette solution for whole-cell patch recording contained (in mmol/L): 120 KCl, 30 NaCl, 1 MgCl2, 0.5 CaCl2, 5 ethylene glycol-bis-(2-aminoethylether)-tetraacetic acid (EGTA), 2 Mg-ATP, and 10 HEPES. The pH of the internal solution was adjusted to 7.2 with KOH. The pipette solution for recording Na+ currents contained (in mmol/L): 120 CsCl, 20 TEACl, 2 MgCl2, 10 EGTA, 2 Mg-ATP, and 10 HEPES. The pH of the internal solution was adjusted to 7.2 with CsOH. The pipette solution for recording transient outward K+ currents contained (in mmol/L): 70 KCl, 1 CaCl2, 2 MgCl2, 70 KF, 10 EGTA, 2 Mg-ATP, and 10 HEPES. The pH of the internal solution was adjusted to 7.2 with KOH. The pipette solution for recording the current-evoked firing of action potentials contained (in mmol/L): 135 K-gluconate, 15 KCl, 5 NaCl, 0.5 EGTA, 10 HEPES, and 2 Mg-ATP. The pH of the internal pipette solution was adjusted to 7.2 with KOH.
The drugs used in this study were all purchased from Sigma, unless otherwise specified. The drugs were first dissolved in ion-free water and then diluted to the final concentrations in the standard external solution just before use. The drugs were applied throughout the experiments with a rapid application technique termed the “Y-tube” method[23] (Figure 1C). The Y-tube was made from polyethylene, and its outlet tip (0.1 mm diameter) was approximately 0.2 mm away from the cell being recorded. The drugs were drawn through the arms of the Y-tube with gentle suction. Some bath fluid was drawn in through the stem so that no agent was applied unintentionally. The system allows for a complete exchange of external solution surrounding a neuron within 20 ms.
Electrophysiological recordings A patch-clamp amplifier (Axopatch 200B; Axon Instruments, Union City, CA, USA) was used for the whole-cell voltage-clamp recordings. Data were sampled and analyzed using a Digidata 1320A interface and a computer installed with Clampex and Clampfit software (version 9.0.1; Axon Instruments, USA). Patch pipettes were pulled from glass capillaries with an outer diameter of 1.5 mm on a 2-stage puller (PP-830; Narishige, Tokyo, Japan). The resistance between the recording electrode filled with pipette solution and the reference electrode was 2–6 MΩ. In most experiments, 70%–90% series resistance compensation was applied, and the membrane potential was held at –60 mV. When recording Na+ currents, data were filtered at 2 kHz and digitized at 10 kHz with membrane potential holding at –80 mV. Fast and slow capacitances were also neutralized. Data were sampled at 5 kHz and filtered by a low-pass Bessel filter set at 1 kHz when recording K+ currents. In addition, currents were corrected for leak and residual capacitance transients by a P/4 protocol. All of the experiments were carried out at room temperature (22–25 °C).
Data analysis All data are shown as the mean±SEM, with statistical significance assessed by Student’s t-test. Statistically significant differences were assumed to be P<0.05 and P<0.01 for all data. P and n represent the value of significance and the number of neurons tested, respectively.
Results
Lidocaine reversibly inhibited the INa Under voltage-clamp conditions, the INa was recorded in cultured rat IC neurons, which were held at –80 mV and depolarized to –30 mV for 20 ms (Figure 2A, left panel). This current was completely and reversibly blocked by 0.3 µmol/L TTX, suggesting that it was mediated by TTX-sensitive, voltage-gated sodium channels. To investigate the effects of lidocaine on the INa, all tested neurons were treated with lidocaine for approximately 30 s before assessing. Lidocaine at 100 µmol/L reduced the INa amplitude to 87.66%±2.12% of that of the control level (n=6, P<0.01; Figure 2A, right panel). After washout, the INa could be recovered to 98.91%±2.19% of that of the control level, indicating a reversible effect of lidocaine on the INa. Moreover, the INa was reduced by lidocaine to a larger extent as the drug concentration was increased from 0.03 to 1 mmol/L (Figure 2A, right panel).
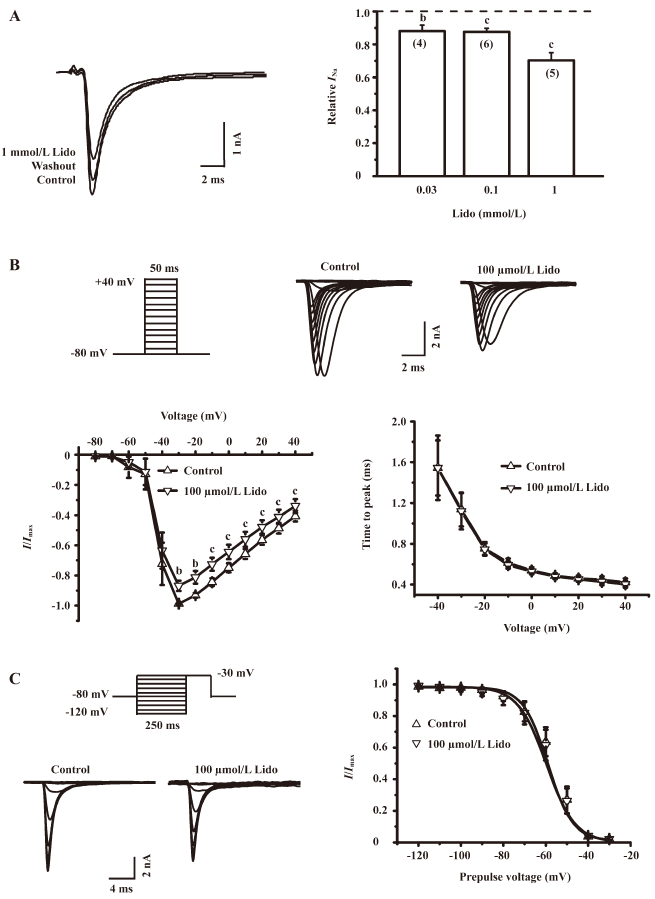
The effect of lidocaine on the current-voltage curve of voltage-gated sodium channels was evaluated. The INa was evoked by step pulses (50 ms wide) from –80 to +40 mV in 10 mV increments (Figure 2B, upper panel). The sodium current was normalized to the maximal current and plotted against evoking potentials. Each of the curves was fitted with a Boltzmann function (Figure 2B, lower left panel). The application of 100 µmol/L lidocaine reduced the peak amplitude of the INa without any significant left or right shift in the current-voltage curve of the INa (n=5, P>0.05). In addition, the application of 100 µmol/L lidocaine did not affect the activation time of the INa (n=7, P>0.05), which is defined as the time between the onset of evoking potentials and the peak amplitude of the INa (Figure 2B, lower right panel).
To further investigate whether lidocaine could also affect the inactivation of voltage-gated sodium channels, we applied step pulses from –120 to –30 mV in 10 mV increments to IC neurons (Figure 2C, left panel). The INa was normalized to the amplitude of the maximal current, and points were fitted with a Boltzmann function. The value of V1/2, at which the conductance of the INa reaches half its maximum, was not significantly shifted by the presence of 100 µmol/L lidocaine (–56.03±0.74 vs –58.97±1.44 mV, n=8, P>0.05; Figure 2C, right panel), indicating that lidocaine does not significantly change the inactivation properties of voltage-gated sodium channels.
Lidocaine reversibly inhibited the IA At a holding potential of –70 mV, the IA of IC neurons was elicited by stepping the membrane potential to +50 mV for 200 ms after a –110 mV prepulse (Figure 3A, left panel). The extracellular application of 0.1, 0.3, and 1 mmol/L lidocaine significantly reduced the amplitude of IA to 96.33%±0.35%, 91.36%±1.64%, and 62.80%±2.61% of that of the control level (n=5, P<0.05), respectively (Figure 3A, right panel). The effects could be removed by the washout of lidocaine for 1 min, indicating that the lidocaine-induced reduction in IA is reversibly inhibited.
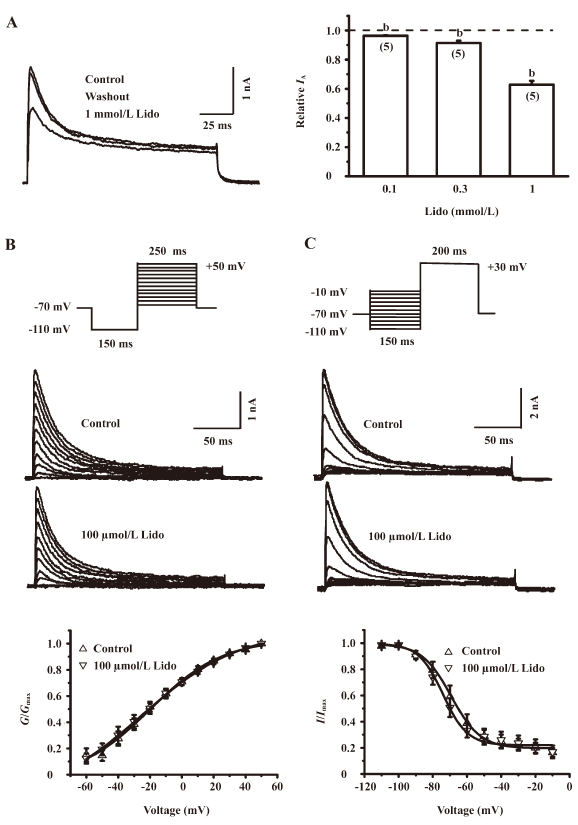
To obtain the conductance-voltage curves, we recorded the IA by using 250 ms depolarizing voltage pulses with 10 mV increments from –60 to +50 mV following a 150 ms prepulse at –110 mV (Figure 3B, upper panel). The IA was converted into conductance by using the equation G=I/(V–VK), where V refers to the evoking potential, and VK refers to the potassium equilibrium potential. The conductance was then normalized and plotted against the membrane potential. Each of the curves was fitted with a Boltzmann function: G/Gmax=(1+exp[V−V1/2]/k)−1, where G is the conductance, Gmax is the maximum conductance, V is the command potential, V1/2 is the potential for half-maximal activation, and k is the slope factor. In the absence of lidocaine, the IA was shown to start activation from approximately –60 mV, and its peak amplitude increased with depolarizing pulse steps. This activation pattern was not significantly influenced by 100 µmol/L lidocaine (Figure 3B, middle panel). In addition, lidocaine at 100 µmol/L also did not significantly change the V1/2 (n=7, P>0.05; Figure 3B, lower panel). These results indicate that lidocaine does not change the steady-state activation of the IA.
To investigate the effects of lidocaine on the steady-state inactivation properties of the IA, we applied a 200 ms test command pulse of +30 mV following 150 ms hyperpolarizing prepulses from –110 to –10 mV in 10 mV increments to IC neurons (Figure 3C, upper panel). The IA recorded with this protocol was normalized and fitted with a Boltzmann function. In the absence of lidocaine, the IA was shown to start inactivation from approximately –100 mV and decrease with hyperpolarizing pulse steps. This inactivation pattern was not significantly influenced by 100 µmol/L lidocaine (Figure 3C, middle panel). In addition, lidocaine at 100 µmol/L also did not significantly shift the V1/2 of the IA inactivation curve (–69.36±2.14 vs –74.08±1.79 mV, n=7, P>0.05; Figure 3C, lower panel). These results indicate that lidocaine does not change the steady-state inactivation of the IA.
Lidocaine reversibly inhibited the IGly Under the condition of voltage-clamp at a holding potential of –60 mV, the extracellular application of 100 µmol/L glycine induced an inward current in all tested neurons, and this current could be almost completely blocked by strychnine, a specific antagonist of the glycine receptor (data not shown). Figure 4A (left panel) shows sample traces of the IGly in the absence and presence of lidocaine at various concentrations. Lidocaine itself at different concentrations did not evoke any detectable current in these neurons; however, lidocaine at 0.1 and 1 mmol/L slightly but significantly reduced the IGly to 91.46%±2.69% and 89.11%±3.17% of that of the control level, respectively (n=5, P<0.05). Lidocaine appeared to suppress the IGly to a larger extent at a higher concentration (3 mmol/L; Figure 4A, right panel). The inhibitory effect of lidocaine on the IGly could recover after washout (Figure 4A, left panel).
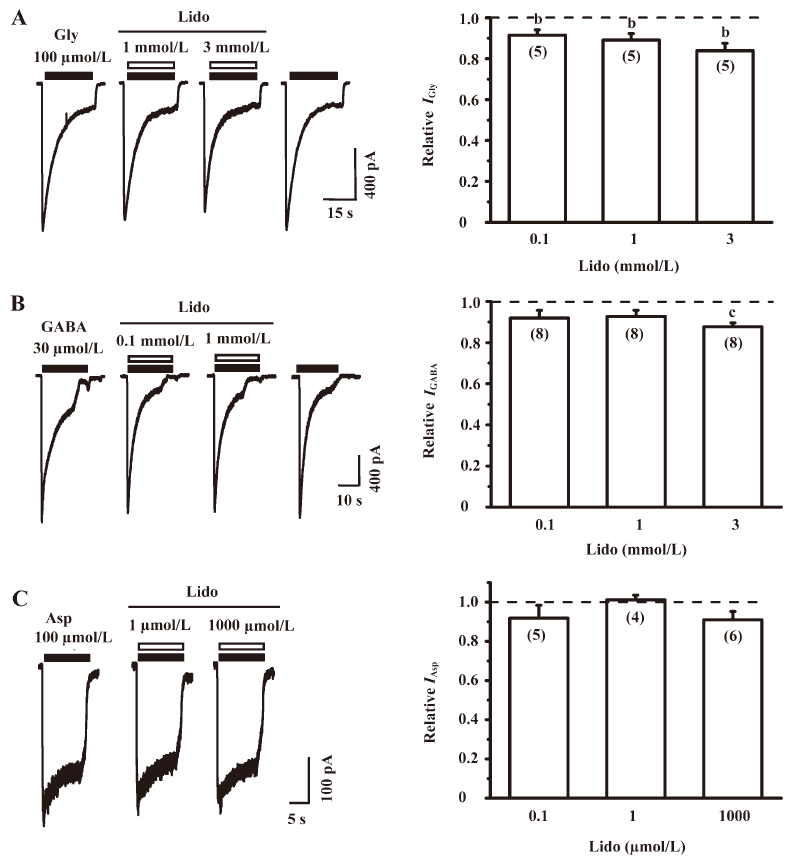
Lidocaine did not affect the IGABA at lower concentrations To study the effect of lidocaine on the IGABA, we recorded the currents induced by 30 µmol/L GABA in the absence and presence of lidocaine at 3 different concentrations (0.1, 1, and 3 mmol/L; Figure 4B, left panel). At a holding potential of –60 mV, the extracellular application of GABA induced an inward current in all tested neurons, and this current could be completely blocked by 10 µmol/L bicuculline, a specific antagonist of GABAA receptors (data not shown), indicating that the IGABA recorded in the present study was mainly mediated by GABAA receptors. Lidocaine at lower concentrations (0.1 and 1 mmol/L) did not have any significant effect on the IGABA, but slightly and significantly reduced the IGABA to 87.70%±1.87% of that of the control level (n=8, P<0.01) at a higher concentration (3 mmol/L; Figure 4B, right panel).
Lidocaine did not affect the IAsp At the holding potential of –40 mV, 100 µmol/L Asp could activate N-methyl-D-aspartic acid (NMDA) receptors in the Mg2+-free extracellular solution containing 1 µmol/L glycine to induce an inward current (Figure 4C, left panel). The IAsp could be completely blocked by 50 µmol/L 2-amino-5-phosphonovaleric acid (APV; a specific NMDA receptor antagonist), indicating that the IAsp recorded in the present study was mainly mediated by NMDA receptors. Lidocaine at 0.1, 1, and 1000 µmol/L did not significantly change the IAsp (n=4−6, P>0.05; Figure 4C, right panel), indicating that lidocaine does not affect NMDA receptors in IC neurons.
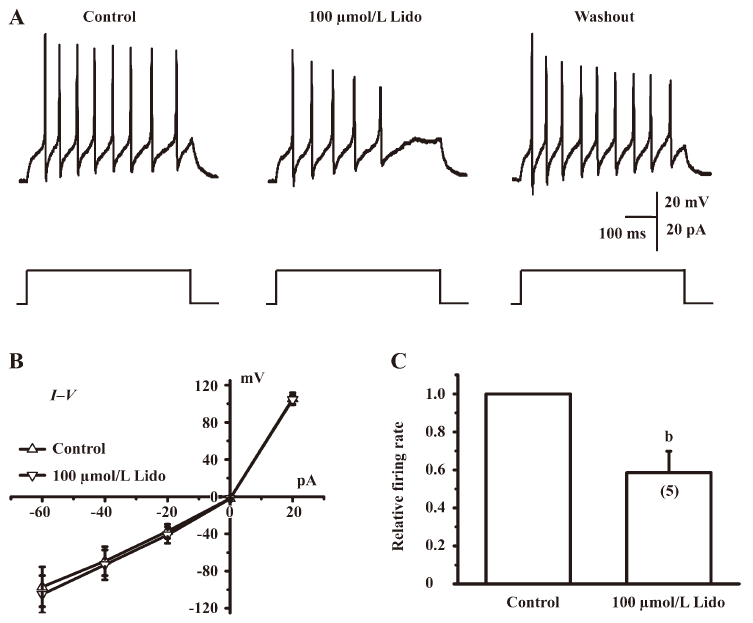
Lidocaine depressed the current-evoked firing of action potentials Because lidocaine had complex and non-specific actions on ion channels and receptors, as shown by the present experiments (Figures 2−4), we sought to determine whether these effects of lidocaine would increase or decrease the excitability of cultured IC neurons. To do so, we recorded the firing rate of action potentials evoked by injecting a positive electrical current in the absence and presence of 100 µmol/L lidocaine under a current-clamp mode. Only neurons that had a resting membrane potential lower than –50 mV (–57.30±1.30 mV, n=5) and did not fire spontaneously were selected for this experiment. Usually action potentials can be evoked by a 20 pA depolarizing current (Figure 5A), and the mean firing rate of recorded neurons is 10.20±3.07 Hz (n=5). Lidocaine at 100 µmol/L did not change the current–voltage relationship (Figure 5B), but significantly depressed the firing rate of action potentials in IC neurons to 58.62%±11.22% of that of the control level (n=5, P<0.05; Figure 5A and C).
Discussion
In the present study, we investigated the effects of lidocaine on major types of ion channels and receptors in IC neurons of the auditory system, including voltage-gated sodium channels, transient outward potassium channels, glycine receptors, GABAA receptors, and NMDA receptors. In addition, we examined whether lidocaine influenced the neuronal excitability by current-evoked firing of action potentials in IC neurons. Lidocaine at concentrations lower than 1 mmol/L significantly suppressed the INa, IA, and IGly to 87.66%±2.12%, 96.33%±0.35%, and 91.46%±2.69% of that of the control level, respectively (Figures 2A, 3A, 4A). At a higher concentration (1 mmol/L), lidocaine further suppressed the 3 currents to 70.26%±4.69%, 62.8%±2.61%, and 89.11%±3.17% of that of the control level, respectively (Figure 2A, 3A, 4A). However, lidocaine at concentrations lower than 1 mmol/L did not significantly affect the IGABA (Figure 4B) and the IAsp (Figure 4C), but slightly depressed the IGABA to 87.70%±1.87% of that of the control level at a higher concentration (3 mmol/L) (Figure 4B). These results demonstrate that the effects of lidocaine on ion channels and receptors in the central auditory system are complex and non-specific. However, lidocaine at 100 µmol/L significantly suppressed the current-evoked firing of action potentials to 58.62%±11.22% of that of the control level (Figure 5), indicating that lidocaine seems to reduce the overall excitability of IC neurons. We suggest that lidocaine at low concentrations (30–100 µmol/L, clinically relevant concentration) reduces the excitability of IC neurons to suppress tinnitus, probably through the inhibition of voltage-gated sodium channels. Alternatively, lidocaine may act on other types of ion channels, receptors[24–26], or synaptic transmissions[27], which were not examined in the present study, to suppress tinnitus.
Effects of lidocaine on voltage-gated channels Voltage-gated sodium channels are essential for action potential generation and its propagation, since they contribute to the rapid rising phase and initial falling phase of action potentials. Therefore, voltage-gated sodium channels play an important role in regulating the excitability of neurons. Voltage-gated sodium channels have 3 distinct states: closed or resting, open, and inactivated. The conformational state in which these channels reside depends on the transmembrane potential[28,29]. It is generally believed that the blockade effect of lidocaine on sodium channels may result from the drug’s interaction with the open state or the inactivated state of the channel[30,31]. Consistent with previous findings, our results show that lidocaine can block the voltage-gated sodium channel, although the inhibition induced by lidocaine is less than that reported by others[30,32]. However, our study demonstrates that lidocaine has no effects on the voltage dependence of the activation state and the time to peak, suggesting that the drug may not interact with the S4 segments, which act as a voltage sensor to initiate activation in response to depolarization signals[33].
Potassium channels are essential for controlling electrical properties and the excitability of neurons, including setting the resting membrane potential, action potential duration, and firing patterns[34]. They also play a vital role in processing auditory information in auditory brainstem nuclei[35]. The transient outward potassium channel has been identified in IC neurons[36], and changes in the amplitude or kinetics of the IA will alter neuronal electrical properties, such as spike repolarization and action potential firing, and influence neuronal excitability[37]. In the present study, lidocaine did not change the amplitude of the IA at a low concentration (100 µmol/L, clinically relevant concentration; Figure 3A), although it significantly reduced the amplitude of the IA at a high concentrations (1 mmol/L, too high to be considered a clinically relevant concentration; Figure 3A, right panel). In addition, lidocaine at a lower concentration did not change its steady-state activation and inactivation (Figure 3B, 3C). Thus, at a clinical dose used for tinnitus treatment, transient outward potassium channels may not be involved in producing suppressive effects on tinnitus. The depression of the IA at a higher concentration (1 mmol/L)may contribute to increase neuronal excitability and can cause neurotoxicity.
Effects of lidocaine on ligand-gated ion channels As major inhibitory neurotransmitters in the mammalian central nervous system, glycine and GABA play an important role in processing auditory information. Glutamate is a major excitatory neurotransmitter and plays an important role in processing auditory information as well. In the present study, lidocaine concentrations lower than 100 µmol/L were found to inhibit the IGly (Figure 4A), but not influence the IGABA (Figure 4B) and the IAsp (Figure 4C). Because the IAsp could be completely blocked by APV, a specific NMDA receptor antagonist (data not shown), the IAsp recorded in the present study was mainly mediated by NMDA receptors. Taken together, these results indicate that lidocaine acts on glycine receptors, but not on GABA receptors or NMDA receptors. The degree to which lidocaine suppresses the IGly was small (up to 91.5% of that of the control level at 100 µmol/L; Figure 4A), which suggests that lidocaine at a clinical concentration does not significantly raise the excitability of the auditory system.
Possible neural basis for the suppressive effects of lidocaine on tinnitus One hypothesis suggests that tinnitus is related to an imbalance between inhibitory and excitatory neurotransmissions in the central nervous system, that is, tinnitus is a consequence of overexcitability in the central auditory system[38]. If this theory holds, an effective therapy for tinnitus should be reducing the excitability of the overexcited auditory system. Our data suggest that lidocaine at a low dose in tinnitus treatment may reduce the excitability in the central auditory system by inhibiting voltage-gated sodium channels. Our data also suggest that transient outward potassium channels, GABAA receptors, and NMDA receptors may not be activated by lidocaine at a low dose. The reduced excitability of IC neurons by lidocaine at lower concentrations has been demonstrated by the drug’s suppressive effects on the current-evoked firing of action potentials (Figure 5). However, our data also suggest that lidocaine may also increase the excitability of the auditory system by inhibiting potassium channels and glycine receptors, particularly when lidocaine is at a high dose. This notion is consistent with the observation that lidocaine at a high dose can cause convulsion[39]. Although our data indicate that the effects of lidocaine on the ion channels and receptors are complex and non-specific in the central auditory system, this drug may nevertheless reduce the overall excitability to suppress tinnitus. Alternatively, lidocaine may act on other types of ion channels, receptors[24–26], or synaptic transmissions[27], which were not examined in the present study, to suppress tinnitus. Further experiments, such as differential responses of excitatory and inhibitory neurons to lidocaine, as well as the effects of lidocaine on alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, are required to provide further insight into its therapeutic mechanism for suppressing tinnitus in the auditory system.
References
- Henry JA, Dennis KC, Schechter MA. General review of tinnitus: prevalence, mechanisms, effects, and management. J Speech Lang Hear Res 2005;48:1204-35.
- Lockwood AH, Salvi RJ, Burkard RF. Tinnitus. N Engl J Med 2002;347:904-10.
- Baguley DM, Jones S, Wilkins I, Axon PR, Moffat DA. The inhibitory effect of intravenous lidocaine infusion on tinnitus after translabyrinthine removal of vestibular schwannoma: a double-blind, placebo-controlled, crossover study. Otol Neurotol 2005;26:169-76.
- Barany R. Die Beeinflussung des Ohrensausens durch intravenos injezierte Lokalanasthetika. Acta Otolaryngol 1935;23:201-3.
- Schmidt H, Davis A, Stasche N, Hormann K. The lidocaine test in tinnitus. Determination of its current status. HNO 1994;42:677-84. In German.
- Majumdar B, Mason SM, Gibbin KP. An electrocochleographic study of the effects of lignocaine on patients with tinnitus. Clin Otolaryngol Allied Sci 1983;8:175-80.
- Eggermont JJ. Central tinnitus. Auris Nasus Larynx 2003;30 Suppl:S7-12.
- Mirz F, Pedersen B, Ishizu K, Johannsen P, Ovesen T, Stodkilde-Jorgensen H, et al. Positron emission tomography of cortical centers of tinnitus. Hear Res 1999;134:133-44.
- Simpson JJ, Davies WE. Recent advances in the pharmacological treatment of tinnitus. Trends Pharmacol Sci 1999;20:12-8.
- Kaltenbach JA. The dorsal cochlear nucleus as a participant in the auditory, attentional and emotional components of tinnitus. Hear Res 2006;216–7:224-34.
- Moller AR. Neural plasticity in tinnitus. Prog Brain Res 2006;157:365-72.
- Wang HT, Luo B, Zhou KQ, Xu TL, Chen L. Sodium salicylate reduces inhibitory postsynaptic currents in neurons of rat auditory cortex. Hear Res 2006;215:77-83.
- House JW, Brackmann DE. Tinnitus: surgical treatment. Ciba Found Symp 1981;85:204-16.
- Reyes SA, Salvi RJ, Burkard RF, Coad ML, Wack DS, Galantowicz PJ, et al. Brain imaging of the effects of lidocaine on tinnitus. Hear Res 2002;171:43-50.
- Kalcioglu MT, Bayindir T, Erdem T, Ozturan O. Objective evaluation of the effects of intravenous lidocaine on tinnitus. Hear Res 2005;199:81-8.
- Vermeij P, Hulshof JH. Dose finding of tocainide in the treatment of tinnitus. Int J Clin Pharmacol Ther Toxicol 1986;24:207-12.
- Trellakis S, Benzenberg D, Urban BW, Friederich P. Differential lidocaine sensitivity of human voltage-gated potassium channels relevant to the auditory system. Otol Neurotol 2006;27:117-23.
- Ruth RA, Gal TJ, DiFazio CA, Moscicki JC. Brain-stem auditory-evoked potentials during lidocaine infusion in humans. Arch Otolaryngol 1985;111:799-802.
- Manabe Y, Yoshida S, Saito H, Oka H. Effects of lidocaine on salicylate-induced discharge of neurons in the inferior colliculus of the guinea pig. Hear Res 1997;103:192-8.
- Basta D, Ernst A. Effects of salicylate on spontaneous activity in inferior colliculus brain slices. Neurosci Res 2004;50:237-43.
- Bauer CA, Brozoski TJ, Holder TM, Caspary DM. Effects of chronic salicylate on GABAergic activity in rat inferior colliculus. Hear Res 2000;147:175-82.
- Tang ZQ, Lu YG, Zhou KQ, Xu TL, Chen L. Amiloride attenuates glycine-induced currents in cultured neurons of rat inferior colliculus. Biochem Biophys Res Commun 2006;350:900-4.
- Murase K, Ryu PD, Randic M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett 1989;103:56-63.
- Arias HR. Role of local anesthetics on both cholinergic and serotonergic ionotropic receptors. Neurosci Biobehav Rev 1999;23:817-43.
- Hirota K, Okawa H, Appadu BL, Grandy DK, Lambert DG. Interaction of local anaesthetics with recombinant mu, kappa, and delta-opioid receptors expressed in Chinese hamster ovary cells. Br J Anaesth 2000;85:740-6.
- Yunoki M, Nakahara T, Mitani A, Maruko T, Kubota Y, Sakamoto K, et al. Lidocaine attenuates muscarinic receptor-mediated inhibition of adenylyl cyclase in airway smooth muscle. Eur J Pharmacol 2003;470:65-71.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidocaine. Nature 1966;209:207-8.
- Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol 1952;116:449-72.
- Armstrong CM. Sodium channels and gating currents. Physiol Rev 1981;61:644-83.
- Chevrier P, Vijayaragavan K, Chahine M. Differential modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br J Pharmacol 2004;142:576-84.
- Matsubara T, Clarkson C, Hondeghem L. Lidocaine blocks open and inactivated cardiac sodium channels. Naunyn Schmiedebergs Arch Pharmacol 1987;336:224-31.
- Xiao YF, Ke Q, Wang SY, Yang Y, Chen Y, Wang GK, et al. Electrophysiologic properties of lidocaine, cocaine, and n-3 fatty-acids block of cardiac Na+ channels. Eur J Pharmacol 2004;485:31-41.
- Ptacek LJ, George AL Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, et al. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenital. Neuron 1992;8:891-7.
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res 1990;83:161-87.
- Grigg JJ, Brew HM, Tempel BL. Differential expression of voltage-gated potassium channel genes in auditory nuclei of the mouse brainstem. Hear Res 2000;140:77-90.
- Sivaramakrishnan S, Oliver DL. Distinct K currents result in physiologically distinct cell types in the inferior colliculus of the rat. J Neurosci 2001;21:2861-77.
- Wang Y, Strahlendorf JC, Strahlendorf HK. A transient voltage-dependent outward potassium current in mammalian cerebellar Purkinje cells. Brain Res 1991;567:153-8.
- Eggermont JJ. Tinnitus: neurobiological substrates. Drug Discov Today 2005;10:1283-90.
- Kurt M, Bilge SS, Kukula O, Kesim Y, Celik S. The role of nitrergic system in lidocaine-induced convulsion in the mouse. Jpn J Pharmacol 2001;85:92-4.