
Neuroprotective effect of sodium ferulate and signal transduction mechanisms in the aged rat hippocampus1
Introduction
The hippocampus is a target of age-related physiological and structural changes. Alterations in the hippocampus during aging are paralleled by behavioral and functional deficits in hippocampus-dependent learning and memory tasks[1]. This impairment is associated with inflammatory and oxidative changes. It has recently been reported that there is an increase in hippocampal concentration of the proinflammatory cytokine, interleukin-1β (IL-1β) and an increase in reactive oxygen species production[2,3] in aged rats. A number of cellular responses are shared by IL-1β and reactive oxygen species, which have been shown to activate certain mitogen-activated protein kinases (MAPK)[4,5]. Three subfamilies of these kinases have been clearly identified in mammals: extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 MAPK. Although ERK is stimulated by growth factors and activation results in neurite outgrowth, cell proliferation or differentiation[6], JNK and p38 MAPK are activated by environmental stress including oxidative stress and heat shock[7]. The age-related decrease in ERK1/2 and increase in p38 MAPK and JNK1/2 were observed in hippocampus of aged rats, which in turn impacts on the ability of the aged rats to sustain long-term potentiation (LTP)[8,9].
An increase in stress-induced cell signaling in hippocampus may lead to cell death in the aged brain but it seems reasonable to propose that this may be coupled with a decrease in cell survival signaling pathways. The serine/threonine kinase Akt/protein kinase B (PKB) is activated via a phosphoinositide-3 kinase (PI3K)-dependent signaling pathway when cells or tissues are exposed to growth factors, insulin, and certain cytokines[10]. PI3K is an intracellular signaling enzyme that has been implicated in cell survival[11]. Akt/PKB has received widespread attention as an important anti-apoptotic protein[12].
Sodium ferulate (SF), extracted from a traditional Chinese herbal medicine, has potent antioxidant[13] and anti-inflammatory activities[14]. It has recently been reported that long-term administration of ferulic acid protected mice against learning and memory deficits induced by centrally administered β-amyloid[15]. The primary site of action of ferulic acid could be microglia[16] and astrocytes[17]. It has been recently reported that ferulic acid inhibited formation of Aβ fibrils and destabilized preformed fibrillary Aβ[18]. Sultana et al reported that ferulic acid ethyl ester significantly inhibited Aβ1-42-induced cytoxicity, intracellular reactive oxygen species accumulation, lipid peroxidation, and induction of inducible nitric oxide synthase in primary hippocampal cultures[19]. Significantly, we have recently reported that SF had inhibitory effects on Aβ-induced increases of IL-1β expression and p38 MAPK pathway and apoptosis in rat hippocampus[20,21]. In addition, treatment with SF protected against glutamate-induced apoptosis of cultured cortical neurons[22]. In the present study, we set out to investigate whether SF treatment was effective in reversing age-associated changes in IL-1β and the JNK signaling pathway in the hippocampus of aged rats.
Materials and methods
Sodium ferulate, a colorless powder with purity >99%, was obtained from Suzhou Changtong Chemical Co (Suzhou, China). The enhanced chemiluminescence kit was from Pierce Biotechnology Inc (Rockford, IL, USA). Phospho-mitogen-activated protein kinases kinase (MKK) 4 (Thr261, N
Animal maintenance and drug exposure protocol Male Sprague-Dawley rats (Experimental Animal Center of Liaoning Medical University, Liaoning, China) at mean age of 3 months (210−270 g, young) or 21 months (500−600 g, aged) were used in these experiments. Animals were housed in pairs (aged rats) or groups of 4 (young rats) at an ambient temperature of 22−24 °C under a 12:12 h light:dark cycle and rats were maintained under veterinary supervision throughout the study. Body weights were monitored and animals that demonstrated external evidence of disease or significant loss of body weight were eliminated from the experiment. Drug doses and durations of treatments used for the experiments were based on our published study[21]. The aged animals were subdivided into three groups. Two groups were fed on a diet enriched in SF (100 mg/kg and 200 mg/kg, daily for 4 weeks). The non-drug-treated control group received standard laboratory chow. The groups of young rats (3 months) received SF (100 mg/kg) or standard laboratory diet. At the end of this period rats were 4 (230−300 g) and 22 (550−650 g) months old, respectively. In addition, 21-month-old rats were used for control animals in the Western blot experiments. Feed was prepared freshly each day and food and water intake did not vary between groups. Hippocampal slices (500 µm thick) were prepared and immediately frozen on dry ice. The CA1 region was microdissected as previously described[23] for Western blot (5 rats in each group). Animals (5 in each group) used for Nissl staining and GFAP immunohistochemical staining, and fluorescent double immunostaining were anesthetized and perfused transcardially with 4% paraformaldehyde.
Western blot analysis Western blot analysis was carried out to analyze the expression of IL-1β, phospho-MKK4, phospho-JNK1/2, phospho-c-Jun, phospho-MEK1/2, phospho-ERK1/2, phospho-Akt, phospho-p70S6K caspase-7, and caspase-3. The fresh hippocampal CA1 region was homogenized in RIPA buffer [1% Triton, 0.1% SDS, 0.5% deoxycholate, 1 mmol/L EDTA, 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 10 mmol/L NaF, 1 mmol/L Na3VO4, 0.1 mmol/L PMSF]. The nuclear fractions were first isolated by centrifuging the homogenates at 7500×g for 30 min at 4 °C. The supernatant was further centrifuged at 12 000×g for 20 min at 4 °C to remove insoluble materials. Protein concentrations were quantified by the method of Lowry. Tissue samples were equalized for protein concentration. Proteins were resolved by 10%–12% SDS-PAGE and transferred to nitrocellulose membranes. Gels were also loaded with colored molecular weight markers to assess electrophoretic transfer and biotinylated protein ladder marker to estimate the molecular weights of bands of interest. The membranes were blocked with 3% BSA in TBS (pH 7.6) for 1 h and incubated overnight at 4 °C with suitably diluted primary antibodies. After extensive washing with TTBS, the membranes were incubated with anti-rabbit IgG, HRP-linked antibody and anti-biotin antibody for 1 h at room temperature. The blots were detected using the enhanced chemiluminescence (ECL) reaction. Following Western immunoblotting for phospho-MKK4, phospho-JNK1/2, phospho-c-Jun, phospho-MEK1/2, phospho-ERK1/2, phospho-Akt, and phospho-p70S6K, blots were stripped and reprobed for total MKK4, JNK1/2, c-Jun, MEK1/2, ERK1/2, Akt, and p70S6K. Following Western immunoblotting for IL-1β, blots were stripped and reprobed for β-actin antibody to ensure equal loading of protein. Quantification of protein bands was achieved by densitometric analysis using Chem Image 5500 software (UVP, USA). The ratio of phosphorylated MKK4, JNK1/2, c-Jun, MEK1/2, ERK1/2, Akt, and p70S6K to the total abundance of the respective proteins of young control rats was normalized to 1.
Nissl staining The rats (5 rats of each group) were perfused transcardially with 4% paraformaldehyde in phosphate-buffered saline (PBS). The brains were post-fixed for 24 h and embedded in paraffin. Serial coronal sections (5 µm thickness) were taken from various sections of the brain, stained for Nissl body using cresyl violet, and examined for pathological changes.
To assess hippocampal injury, the number of neurons in the pyramidal layer of the hippocampal CA1 region was counted under a light microscope at 400× magnification according to the method described by Zhang et al[24]. Briefly, two continuous fields in the hippocampal CA1 subregion were selected for each section and the neurons were counted. The mean of the two fields was taken as the neuron number of this section and the mean of four sections was taken as the neuron number of this specimen.
Immunohistochemical staining and double immunofluorescence study Tissue sections were processed for immunohistochemistry and double immunofluorescence study. Endogenous peroxidase activity was blocked by incubation with 3% H2O2 for 10 min. The sections were incubated with 10% normal goat serum. The blocking serum was removed, and sections were incubated with polyclonal antibody against GFAP (1:100 in TBS) overnight at 4 °C, then with biotinylated secondary antibody at 37 °C for 20 min. The GFAP-positive cells were detected by using SABC and DAB kits. The sections for double immunofluorescence labeling were incubated in IL-1β (dilution 1:50) or phospho-JNK (dilution 1:50) antibody with integrin αM (dilution 1:50) or GFAP (dilution 1:50) overnight at 4 °C. After incubation, fluorescein isothiocyanate (FITC) conjugated and Cy3 conjugated secondary antibodies (Santa Cruz, CA, USA) were added after washing in PBS. All images were captured under a Leica TCS-SP2 (Germany) confocal microscope. For quantitative image analysis of hippocampal integrin αM immunostaining, serial sagittal sections of one hemisphere taken from lateral (+0.5 to +2.25) were examined. Integrin αM immunostaining was evaluated on sagittal brain sections of 5 animals from each group. For each animal, antigens were detected in 5 parallel sections having a defined distance of 35 µm showing hippocampus. The number of integrin αM positive cells was counted and is shown as immunopositive cells per mm2.
Statistical analysis All data are presented as mean± SD. Statistical analysis was carried out with one-way ANOVA, followed by least significant difference (LSD) post hoc test, which was provided by SPSS 11.5 statistical software. The level of significance was accepted as P<0.05.
Results
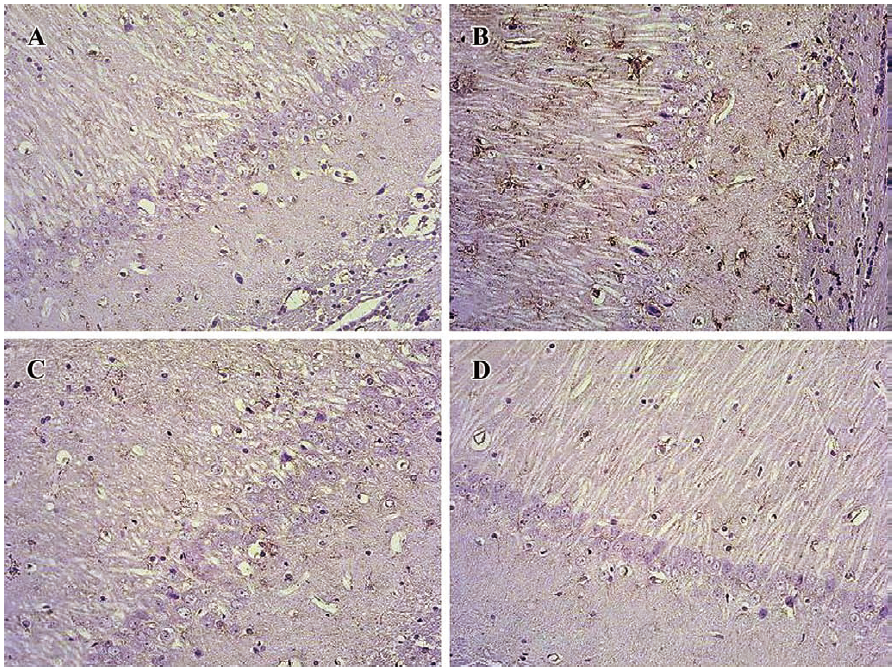
Age-related alteration in IL-1β protein expression and glial activation were reversed by sodium ferulate in the hippocampus As shown in Figure 1A, the basal level of IL-1β protein expression in hippocampus of young, control-treated rats was very low. IL-1β protein expression was significantly higher in hippocampus from 22-month-old control-treated rats compared with young control-treated rats. SF treatment inhibited the age-related increase in IL-1β protein expression, but it did not affect IL-1β expression in hippocampus prepared from young rats. Quantitative counting of microglial cells showed that oral SF treatment effectively reduced the number of microglia in hippocampus of aged rats (Figure 1B). Confocal immunofluorescence for IL-1β and integrin αM (OX-42), a specific marker of microglia, or for IL-1β and GFAP showed that the immunostaining of IL-1β predominantly co-localized with integrin αM (OX-42) in the hippocampus (Figure 1C). The vast majority of IL-1β-positive microglial cells showed a round to oval morphology, indicating an activated state of inflammation in hippocampus of aged rats. However, IL-1β-positive astrocytes were not found in hippocampus of rats (Figure 1D). Astrocytes were visualized by means of immunohistochemistry for GFAP, a specific marker of astrocytes. In aged hippocampus and cortex, a marked infiltration of astrocytes was found as well as transformation of astrocytes from a resting to an activated state, highlighted by phenotypic changes characterized by long, thick branching. Oral SF treatment at doses of 100 mg/kg and 200 mg/kg daily for 4 weeks significantly reduced the age-related astrocytic reaction in hippocampus and cortex (Figure 2). In addition, IL-1β protein expression was significantly enhanced in hippocampus from 21-month-old control rats compared with young 4-month-old control rats (data not shown).

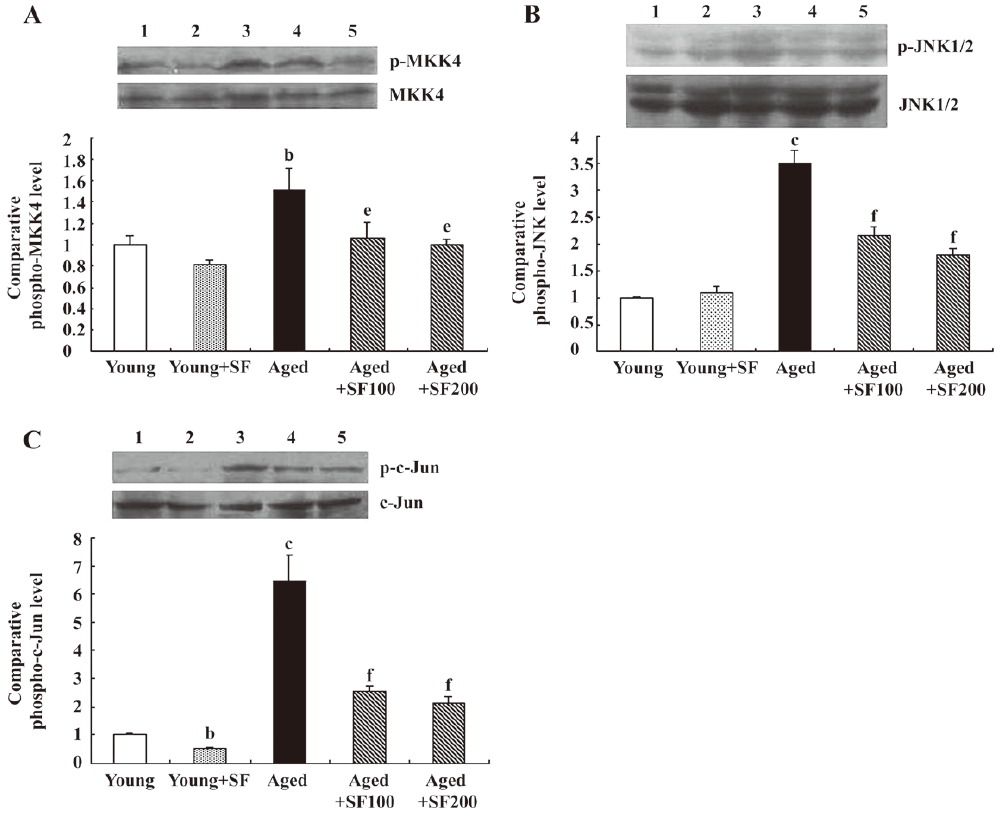
Sodium ferulate inhibited age-related increase in phospho-MKK4, phospho-JNK and phospho-c-Jun expressions A good deal of evidence suggests that one consequence of increased IL-1β in several cell types is activation of JNK and this has been reported in hippocampus[5]. Therefore, hippocampal tissue prepared from young and aged rats was assessed for phosphorylated MKK4 (p-MKK4), phospho-JNK1/2 (p-JNK) and phospho-c-Jun expressions. Figure 3A shows a sample immunoblot indicating that expressions of p-MKK4 (but not total MKK4), which is known to activate JNK, was enhanced in 22-month-old rats compared with young rats. The age-related increase in MKK4 phosphorylation was paralleled by the changes in p-JNK (Figure 3B) and p-c-Jun (Figure 3C); assessment of the data obtained from densitometric analysis indicated that the age-related increase in p-JNK and p-c-Jun was statistically significant, but the expressions of total JNK and c-Jun were not affected by treatment. The expressions of p-MKK4, p-JNK, and p-c-Jun in hippocampus prepared from 21-month-old rats were enhanced compared with that in young control rats (data not shown). SF (100 mg/kg and 200 mg/kg, daily for 4 weeks) treatment abrogated the aged-related increase of p-MKK4, p-JNK, and p-c-Jun. Meanwhile, SF had significant inhibitory effects on p-MKK4, p-JNK, and p-c-Jun expressions in hippocampus prepared from young rats. To determine the cell type that expresses phospho-JNK, a double immunofluorescent study was carried out in the hippocampus of rats. Co-staining of integrin αM and phospho-JNK confirmed that phospho-JNK was predominantly expressed in microglia (Figure 4). In addition, co-staining of GFAP and phospho-JNK showed that phospho-JNK positive astrocytes were small (Figure 4).

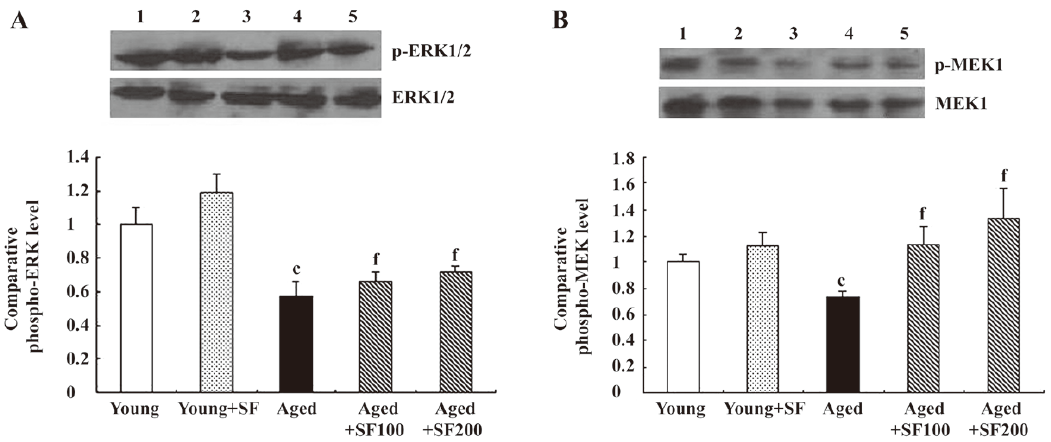
Effects of sodium ferulate on MEK1/2 and ERK1/2 phosphorylation in the hippocampus of aged rats One consistent finding was that ERK activation decreased in the hippocampus and cortex of aged rats[25,26]. This age-related decrease in ERK1/2 is likely to contribute to the age-related deficit in long-term potentiation[27]. Therefore, we analyzed phosphorylated MEK1/2 and ERK1/2 protein expressions. The results revealed that ERK1/2 phosphorylation was decreased in hippocampal preparations obtained from control-treated aged rats compared with control-treated young rats. SF partly inhibited the aged related decrease in p-ERK1/2 (Figure 5A). In parallel with the change of ERK1/2 phosphorylation, we found that p-MEK1/2 expression, an upstream kinase of ERK1/2, was significantly decreased in hippocampus obtained from control-treated aged rats and SF suppressed this change (Figure 5B). Equal protein loading was verified by reprobing immunoblots for total MEK1/2 and ERK1/2 and the data indicate that their expressions were similar in all treatment groups examined.
Sodium ferulate prevented the age-related decreases in phosphorylated Akt/PKB and p70S6K protein expression in the hippocampus of aged rats It has been shown that cell death is accompanied by a decrease in survival signals[28]. We considered that the age-related increase in IL-1β concentration in hippocampus, which is associated with upregulation of phospho-JNK1/2, might be accompanied by an age-related decrease in Akt/p70S6K pathway. Therefore, we analyzed protein expressions of phosphorylated Akt/PKB and p70S6K. The sample immunoblot and mean data in Figure 6 indicated that activation of both kinases was significantly reduced in hippocampus prepared from control-treated aged, compared with control-treated young rats. SF treatment did not significantly affect kinase activation in hippocampus prepared from young rats, but it prevented the age-related decreases so that the values were similar to those in hippocampus prepared from young rats, but the expressions of total Akt/PKB and p70S6K were not affected by treatment (Figure 6A, 6B).

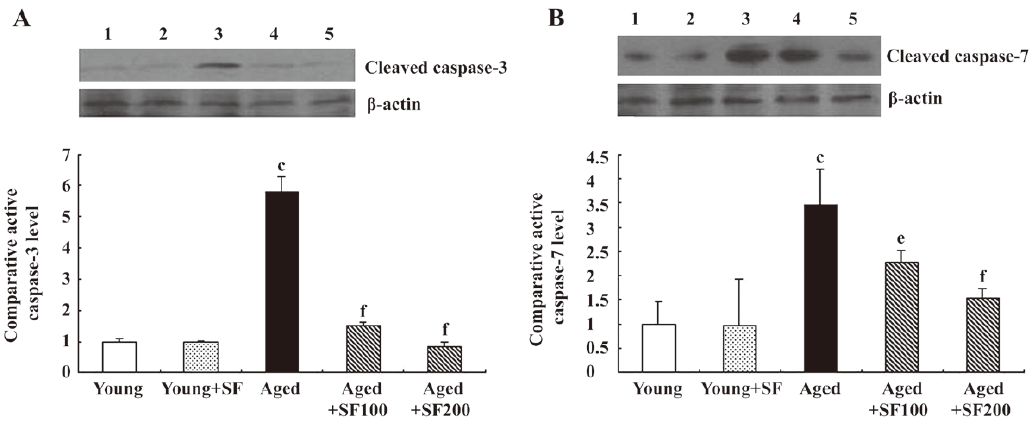
Sodium ferulate inhibited the age-related alterations in caspase-3 and caspase-7 expression in the hippocampus of aged rats The activation of caspase-3 is a hallmark of apoptosis. In the apoptotic pathway, caspase-9 activity is responsible for procaspase-3 and procaspase-7 activation. The activated caspase-3 and caspase-7 of 20 kDa were observed in Western blot analysis. The results showed that expressions of activated caspase-3 and caspase-7 were markedly increased in hippocampus prepared from aged, compared with young, rats. SF treatment prevented this age-related change. Thus, protein expression of activated caspase-3 and caspase-7 in the hippocampus of aged rats treated with SF was similar to that in the hippocampus of young control-treated or SF-treated rats (Figure 7A, 7B).
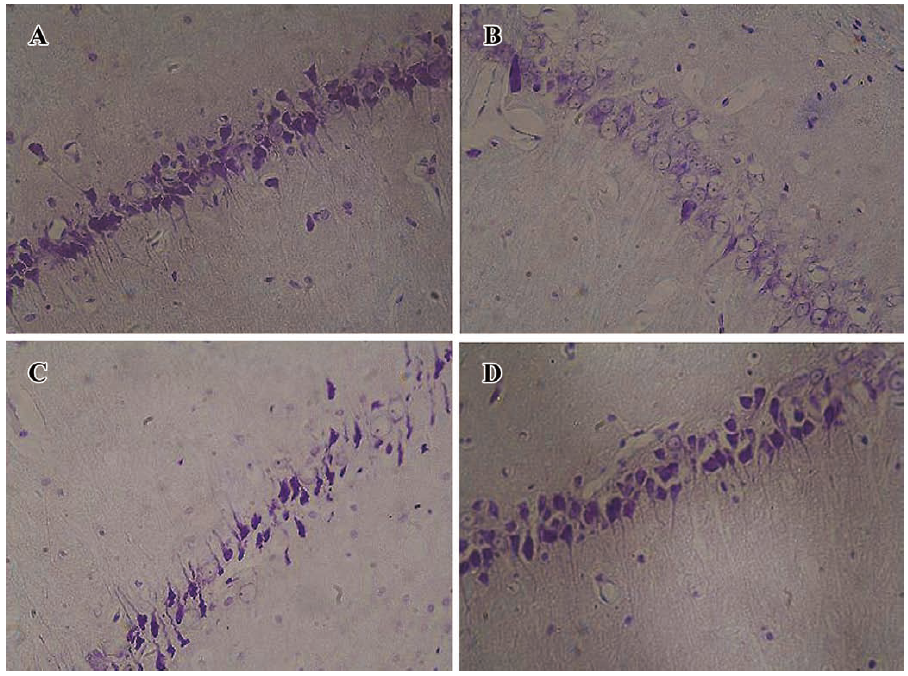
Effect of sodium ferulate on age-related morphological change and number of hippocampal CA1 pyramidal neurons The arrangement of hippocampal CA1 pyramidal neurons of the young-control group is clearly discernible (Figure 8A). The Nissl body in the hippocampal pyramidal neurons of the aged-control group was decreasing or dissolving (Figure 8B). The arrangement of hippocampal pyramidal neurons of SF-treated aged rats was better than that of aged-control rats (Figure 8C, 8D). However, no obvious age-related changes in neuron numbers in hippocampal CA1 regions were found (data not shown).
Discussion
We present evidence demonstrating that the age-related increases in IL-1β expression and JNK activation were accompanied by downregulation of survival signals in the Akt/p70S6K pathway and MEK/ERK pathway. These findings were coupled with increased expression of activated caspase-3 and caspase-7. Significantly, treatment with SF prevented these age-related changes, including the age-related increase in caspase-3 activation, an indicator of cell death. The increase in IL-1β in hippocampus prepared from aged rats, which confirms previous findings[4,9], was coupled with JNK phosphorylation. The increase in JNK phosphorylation is paralleled by several changes that are hallmarks of cell death. For example, phosphorylation of c-Jun in the hippocampus was enhanced, which is a downstream consequence of JNK activation and which has been shown to play a significant role in triggering neuronal apoptosis in a variety of cells in vitro[29,30]. Similarly, increased caspase-3 and caspase-7 activation was observed in hippocampus prepared from aged rats. A recent study linked activation of JNK with translocation of cytochrome c from mitochondria, suggesting that the patency of the mitochondrial membrane was affected by JNK activation[31]. A significant finding of this study is that SF treatment prevented age-related astrocyte activation, increases in MKK4/JNK/c-Jun pathway and caspase-3 activation indicating a potential neuroprotective effect as previously described[20,21]. These findings suggest that the inhibitory effect of SF on the JNK pathway and on caspase-3 activity may be secondary to its ability to suppress the age-related increase in IL-1β.
It has also been reported that cell death is accompanied by a decrease in survival pathways[28]. ERK1/2 plays a role in different types of LTP. Several behavioral studies have also demonstrated the importance of ERK1/2 in learning and memory[32]. Our results showed that, in addition to enhanced JNK and c-Jun activation, the increase in activated caspase-3 expression was associated with attenuated expression of phosphorylated ERK1/2. This observation is in agreement with recent reports showing an age-related decrease in ERK1/2 activity in the hippocampus and cortex of aged rats[9]. ERK1/2 activation and inactivation is regulated by MEK1/2. The age-related decrease in phosphorylated ERK1/2 was paralleled by an age-related decrease in phosphorylated MEK1/2 expression. Significantly, SF treatment prevented the age-related decrease in phosphorylated ERK1/2 and phosphorylated MEK1/2 expressions. Our previous studies showed that SF abolished the Aβ-induced decrease in phosphorylated ERK1/2 in rat hippocampus[20]. It is possible that age-related change in ERK1/2 activation is dependent on MEK1/2 activation. Due to the complexity of the regulation of ERK cascade, it is difficult to delineate mechanisms related to age-associated deficits in this signaling pathway. The activation of the ERK signaling pathway was reported to suppress the proapoptotic activity of JNK, thus protecting rat pheochromocytoma cells (PC-12) from nerve growth factor NGF withdrawal-induced cell death[33]. The MEK/ERK pathway interferes with apoptosis at the level of cytosolic caspase activation, downstream of the release of cytochrome c from mitochondria[34].
It has been reported that increased IL-1β concentration was coupled with downregulation of PI3K in cortical tissue prepared from aged rats[9]. Active PI3K catalyses the synthesis of 3′-phosphorylated inositol lipids that control the intracellular localization and activity of a key molecule of neuronal survival, the protein kinase Akt/PKB. Because the phosphorylation of Akt/PKB serves as an indicator for a previous PI3K activation, we set out to investigate age-related changes in Akt/PKB levels by phosphorylation using phospho-specific antibody. The results showed that Akt/PKB activation decreased with age. Therefore, the age-associate increase in activated caspase-3 may arise from the coupled increase in JNK activation and decrease in ERK and Akt/PKB activation. It has been reported that the anti-inflammatory cytokine IL-4 can lead to activation of PI3K[35]. IL-4 concentration in cortical tissue decreased with age and IL-4 had the capability of increasing the activity of PI3K[9]. A recent study found that IL-4 prevented apoptosis through Akt activation and the p70S6K survival signaling pathway[36]. We considered that an age-related downregulation of Akt may contribute to the decrease of p70S6K. The evidence presented is consistent with that hypothesis. SF treatment reversed the age-associated decreases in cell survival signals Akt and p70S6K.
In conclusion, this study demonstrated that in aged hippocampus, in addition to enhanced JNK/c-Jun pathway, the increases of caspase-3 and caspase-7 were associated with the decrease of the MEK/ERK pathway and the Akt/p70S6K survival signaling pathways. SF treatment prevented these age-related changes, including the increase in caspase-3 activity.
Author contribution
Ying JIN designed research; En-zhi YAN, Xiao-ming LI, Ying FAN performed research; Yan-jie ZHAO, Wan-zhu LIU contributed new analytical reagents and tools; En-zhi YAN analyzed data; Ying JIN wrote the paper.
References
- Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol 2003;69:143-79.
- Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 1998;18:2974-81.
- Murray CA, Clements MP, Lynch MA. Interleukin-1 induces lipid peroxidation and membrane changes in rat hippocampus: An age-related study. Gerontology 1999;45:136-42.
- Martin DS, Lonergan PE, Boland B, Fogarty MP, Brady M, Horrobin DF, . Apoptotic changes in the aged brain are triggered by interleukin-1beta-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J Biol Chem 2002; 277: 34 239−46.
- Vereker E, O’Donnell E, Lynch MA. The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci 2000;20:6811-9.
- Creedon DJ, Johnson EM, Lawrence JC. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem 1996; 271: 20 713−8.
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807-69.
- O’Donnell E, Vereker E, Lynch MA. Age-related impairment in LTP is accompanied by enhanced activity of stress-activated protein kinases: analysis of underlying mechanisms. Eur J Neurosci 2000;12:345-52.
- Maher FO, Martin DS, Lynch MA. Increased IL-1beta in cortex of aged rats is accompanied by downregulation of ERK and PI-3 kinase. Neurobiol Aging 2004;25:795-806.
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997;275:661-5.
- Hawkins PT, Welch H, McGregor A, Eguinoa A, Gobert S, Krugmann S, et al. Signalling via phosphoinositide 3OH kinases. Biochem Soc Trans 1997;25:1147-51.
- Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol 2000;151:483-94.
- Scott BC, Butler J, Halliwell B, Aruoma OI. Evaluation of the antioxidant action of ferulic acid and catechins. Free Radic Res Commun 1993;19:241-53.
- Fernandez MA, Saenz MT, Garcia MD. Anti-inflammatory activity in rats and mice of phenolic acids isolated from Scrophularia frutescens. J Pharm Pharmacol 1998;50:1183-6.
- Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, et al. Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol 2001;133:89-96.
- Kim HS, Cho JY, Kim DH, Yan JJ, Lee HK, Suh HW, et al. Inhibitory effects of long-term administration of ferulic acid on microglial activation induced by intracerebroventricular injection of β-amyloid peptide (1-42) in mice. Biol Pharm Bull 2004;27:120-1.
- Cho JY, Kim HS, Kim DH, Yan JJ, Suh HW, Song DK. Inhibitory effects of long-term administration of ferulic acid on astrocyte activation induced by intracerebroventricular injection of beta-amyloid peptide(1–42) in mice. Prog Neuropsychopharmacol Biol Psychiatry 2005;29:901-7.
- Ono K, Hirohata M, Yamada M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem Biophys Res Commun 2005;336:444-9.
- Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta- peptide(1–42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem 2005;92:749-58.
- Jin Y, Yan EZ, Fan Y, Zong ZH, Qi ZM, Li Z. Sodium ferulate prevents amyloid-beta-induced neurotoxicity through suppression of p38 MAPK and upregulation of ERK-1/2 and Akt/protein kinase B in rat hippocampus. Acta Pharmacol Sin 2005;26:943-51.
- Jin Y, Fan Y, Yan EZ, Liu Z, Zong ZH, Qi ZM. Effects of sodium ferulate on amyloid-beta-induced MKK3/MKK6-p38 MAPK-Hsp27 signal pathway and apoptosis in rat hippocampus. Acta Pharmacol Sin 2006;27:1309-16.
- Jin Y, Yan EZ, Fan Y, Guo XL, Zhao YJ, Zong ZH, et al. Neuroprotection by sodium ferulate against glutamate-induced apoptosis is mediated by ERK and PI3 kinase pathways. Acta Pharmacol Sin 2007;28:1881-90.
- Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, Morrison JH, et al. Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of Ca2+/calmodulin-dependent protein kinase II in long-term potentiation. J Neurosci 2001;21:7053-62.
- Zhang YM, Yang Q, Xu CT, Li KS, Li WQ. Effects of phenytoin on morphology and structure of hippocampal CA3 pyramidal neurons of rats in chronic stress. Acta Pharmacol Sin 2003;24:403-7.
- Hu Y, Schett G, Zou Y, Dietrich H, Xu Q. Abundance of platelet-derived growth factors (PDGFs), PDGF receptors and activation of mitogen-activated protein kinases in brain decline with age. Brain Res Mol Brain Res 1998;53:252-9.
- Ikeyama S, Kokkonen G, Shack S, Wang XT, Holbrook NJ. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J 2002;16:114-6.
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 1997; 272: 19 103–6.
- Macdonald NJ, Decorti F, Pappas TC, Taglialatela G. Cytokine/neurotrophin interaction in the aged central nervous system. J Anat 2000;197:543-51.
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, et al. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci 1998;18:5124-35.
- Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol 1999;19:751-63.
- Lonergan PE, Martin DS, Horrobin DF, Lynch MA. Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J Biol Chem 2002; 277: 20 804–11.
- Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 2002;42:135-63.
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995;270:1326-31.
- Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol 1999;19:5308-15.
- Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999;17:701-38.
- Lin SJ, Chang C, Ng AK, Wang SH, Li JJ, Hu CP. Prevention of TGF-beta-induced apoptosis by interlukin-4 through Akt activation and p70S6K survival signaling pathways. Apoptosis 2007;12:1659-70.