
Mitochondria-dependent apoptosis induced by a novel amphipathic photochemotherapeutic agent ZnPcS2P2 in HL60 cells1
Introduction
Photodynamic therapy (PDT), a promising cancer therapeutic approach, utilizes a photosensitizing agent followed by illumination at a specific “activating” wavelength of light. PDT produces singlet oxygen and other reactive oxygen species (ROS), leading to lipid peroxidation and damage to membranes, DNA, cytoskeleton, and other sites, and eventual cell death[1–3]. Individually, photosensitizers and light are nontoxic, but tumoricidal when combined. Electron microscopy, histological and biochemical studies have shown that PDT with different photosensitizers induces apoptotic or necrotic cell death in different cell types[4–8]. Photosensitizers are key factors for PDT. According to previous studies[9,10], ZnPcS2P2 presents 4 advantages over conventional photosensitizers such as hematoporphyrin derivatives (HpD) or other porphyrin derivatives. The first advantage is that the wavelength of suitable exicited light for ZnPcS2P2 is 670 nm, which is good for penetrating into tissues. The second advantage is that ZnPcS2P2 has no obvious absorption in the visible part as with HpD with the result of decreased skin phototoxicity from natural light to a larger extent. The third advantage is that the exicited triplet state of ZnPcS2P2 has a larger quantum yield and longer lifetime. The fourth advantage is that the amphipathic structure of ZnPcS2P2 results in the increase of selective uptake of sensitizers by tumor cells. Our previous investigation manifested that leukemic cell lines exhibited higher susceptibility to ZnPc-PDT than normal hematopoietic cells and ZnPc-PDT could eliminate the residual leukemia cells in normal bone marrow mononuclear cells[11]. However, the mechanism underlying the killing effects of ZnPc-PDT on leukemic cells is still unknown. In the present paper, we studied the death pathway of HL60 cells induced by ZnPc-PDT and the mechanism of its action.
The mitochondrion plays a central role in the control of apoptosis by releasing the apoptogenic proteins apoptosis-inducing factor (AIF) and cytochrome c into the cytosol. The released proteins activate pathways essential for carrying out the morphological and biochemical changes. For example, cytochrome c, normally residing between the mitochondrial outer and inner membranes and serving as a diffusible electon carrier in the intermembrane space, has been demonstrated to be a co-activator of caspase-9, which in turn activates caspase-3. The active caspase-3 then activates caspase-activated DNA (CAD). Active CAD initiates chromatin and DNA fragmentation[12–22]. Therefore, in the present study, we investigate the relationship between the death mechanism induced by ZnPc-PDT and the mitochondria-dependent apoptosis. Furthermore, it is well known that Bcl-2 serves to inhibit apoptosis by antagonizing the release of mitochondrial cytochrome c, whereas Bax promotes apoptosis[24,25]. The Bcl-2 protein was reported to be a damage target of PDT and contributes to PDT-efficient induction of apoptosis[25]. Therefore, we also detect the kinetic change of Bcl-2 and Bax expression following ZnPc-PDT.
Materials and methods
Cell line Human myelogenous leukemia HL60 cells provided by the Shanghai Institute of Cytobiology (Institute of Chinese Academy of Sciences, China) were maintained in continuous culture in RPMI-1640 (GIBCO BRL), supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS), penicillin 100 U/mL, 100 µg/mL streptomycin, and 0.25 mmol/L L-glutamine at 37 °C under 5% CO2 humidified air. The viability of cells was determined by Trypan blue dye exclusion. Cells were maintained in log phase with viability greater than 95%.
Photosensitizer ZnPcS2P2 was a gift from the Department of Chemistry, Institute of Research on Functional Materials, Fuzhou University, China. It was dissolved in a solution comprised of Cremophor EL 2%(v/v), propylene glycol 20% (v/v), NaCl 0.9%(w/w) and stored in the dark at 4 °C.
Photodynamic treatment HL60 cell suspensions (5×106/mL in RPMI-1640) were incubated with 0.5 µmol/L of ZnPcS2P2 for 5 h. The cell suspensions were then photo-irradiated using a LD-670 semi-conductor laser (Laboratory of Laser Medicine, Tianjin Medical University, China) emitting red light at a 670 nm wavelength. The power density at the illumination area was 53 mW/cm2 and total light dose was 2.1 J/ cm2. A control group (cells incubated with same volume of saline for 5 h) was established. Thereafter, cells were harvested at 6 h, 12 h, and 24 h, respectively.
Apoptosis assays Ultrastructure was analyzed under a HU-12A electron microscope. DNA fragmentation assay was followed as described[26]. TUNEL stains were carried out according to the manufacture’s instructions (Promega) and the apoptotic cells characterized with brown nuclei were counted under microscope and photographed.
Assays for mitochondrial membrane potential (Δψm), caspase-family protease activity and caspase-3 activity The procedure was carried out according to the manufacture’s instructions (Biovision). The results were then analyzed by flow cytometry (BD). The Δψm detection kit utilized MitoCaptureTM (BioVision), a cationic dye that fluoresces differently in healthy cells and apoptotic cells. In healthy cells, MitoCapture accumulated and aggregated in the mitochondria, giving off a bright red fluorescence. In apoptotic cells, MitoCapture could not aggregate in the mitochondria due to the altered Δψm, and thus it remained in the cytoplasm in its monomer form, emitting green fluores-cence. The fluorescent signal could be analyzed by flow cytometry FITC channel for green monomers and PI channel for red aggregates. The assay for caspase activity was based on the cleavage of (aspartyl) 2-Rhodamine 110 (D2R), a reported substrate for members of caspase family proteases. D2R was non-fluorescent, however, upon cleavage of the substrate by cellular caspases, the released rhodamine 110 resulted in a green fluorescence. The caspase-3 activity detection kit utilized the caspase-3 inhibitor DEVD-FMK, conjugated to FITC as the fluorescent in situ marker. FITC-DEVD-FMK was cell-permeable, nontoxic, and irreversibly binds to activated caspase-3 in apoptotic cells.
Assay for cytochrome c Western blotting was performed according to previously published methods[27], with minor modification. In brief, cell pellets were washed once with ice-cold phosphate-buffered saline (PBS) and lysed in lysis buffer (in mmol/L: 20 Hepes-KOH, 250 sucrose, pH 7.5, 10 KCl, 1.5 MgCl2, 1 sodium ethylene diaminetetraacetic acid (EDTA), 1 dithiothreitol, and 0.1 phenylmethylsulfonyl fluoride). The cells were homogenized on ice for 15 min and the homogenates were centrifuged twice at 1000×g for 10 min. The supernatants were centrifuged at 12000×g for 15 min at 4 °C, and the supernatants were cytosol without mitochondria. Protein concentration of cell extracts was determined with Bradford protein assay (Coomassie Brilliant Blue G-250 obtained from BBI). Cytosol protein was loaded and separated on 12% sodium dodecyl sulfate (SDS) polyacrylamide gel and transferred to a nitrocellulose membrane by standard electric transfer protocol. The chemiluminescent detection of membrane-bound proteins was performed according to the manufacture’s instructions (KPL, Kirke-gaard & Perry Laboratories). In brief, the membranes were blocked in blocking buffer for 1 h at room temperature and then incubated with anti-human cytochrome c mouse monoclonal antibody (Neomarkers) in blocking buffer overnight at 4 °C. After washing, the membranes were incubated with anti-mouse secondary antibody horseradish peroxidase conjugate and then detected by enhanced chemiluminescent detection system. Finally, the bands were visualized by autoradiography using X-ray film and scanned and quantified by Imager Quant (Alpha Innotech Corporation, San Leandro, USA). The relative levels of cytochrome c were obtained after normalization with β-actin values in the same lane. At the same time, the distribution of cytochrome c was detected by immunohistochemistry staining and the signals were visualized with an alkaline phosphatase-linked assay system (DAKO LSAB labeled streptavidin-biotin, DakoCytomation Inc). The proteins were detected under microscope and photographed.
Assay for Bcl-2 protein and Bax protein To prepare lysates, cells were initially washed once with ice-cold PBS. Cell pellets were treated with lysis buffer [1% Nonidet P-40 detergent (NP-40), 50 mmol/L Tris-HCl buffer, pH 8, 0.1% SDS, 0.1 mmol/L phenylmethylsulphonyl fluoride (PMSF), 0.02% sodium azide] for 15 min on ice. Lysates were centrifuged at 12 000×g 4 °C for 15 min. Protein concentration of cell extracts was determined with Bradford protein assay. The detergent-soluble proteins were detected using the same procedure as cytochrome c. The intensities of the bands were determined with the utilization of the ratios of Bax (21 kDa) to Bcl-2 (26 kDa). The anti-human monoclonal antibodies were purchased from NeoMarkers.
Statistical analysis All data were presented as mean±SD of 3 independent experiments. Statistical significance was determined with Student’s unpaired two-tailed t-test (SPSS 10.0 software, SPSS Inc, Chicago, USA). P<0.05 was considered statistically significant.
Results
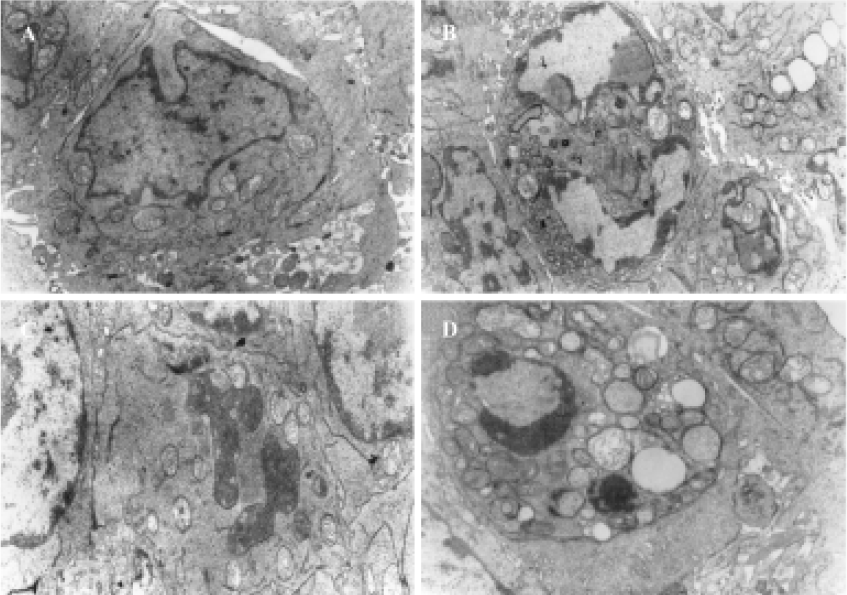
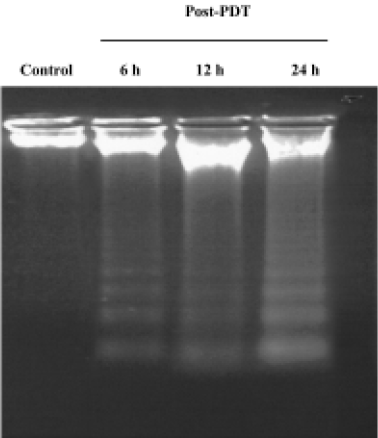
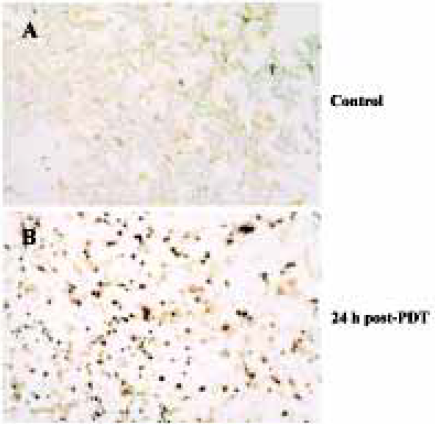
Characteristics of apoptosis in HL60 cells after ZnPc-PDT Electron microscopy studies showed that in ZnPc-PDT group, the features of HL60 cells appeared obviously as following: the cell size decreasing, cell membrane blebbing, cytoplasm shrinkage, nuclear chromatin condensation or cleavage and formation of apoptotic bodies (Figure 1). And characteristic chromosomal DNA fragmentation appeared in the cells after ZnPc-PDT. By contrast, no DNA ladder existed in the control groups (Figure 2). TUNEL staining analysis showed many apoptotic cells which nuclei stained brown existed in ZnPc-PDT groups and the average apoptosis rates significantly increased in a time-dependent manner. At the 3 different time points after PDT, the mean TUNEL-positive rates were 9.6%, 24.4%, and 33.0%, respectively (Figure 3). However, few apoptotic cell was detected in the control group. The results indicated that ZnPc-PDT could induce evident apoptosis in HL60 cells.
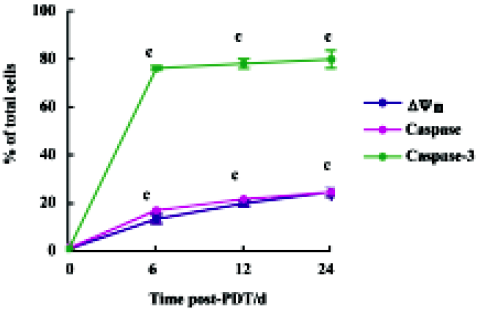
Characteristics of mitochondria-dependent apoptosis in HL60 cells after ZnPc-PDT In the control group, the positive rates of assays for Δψm, activities of caspase family proteases and caspase-3 were all less than 1%. In the ZnPc-PDT groups, compared with the control group, the positive rates of the 3 indexes increased remarkably (P<0.01) as shown in Figure 4. In the ZnPc-PDT groups, Δψm altered significantly and the positive rates increased in a time-dependent manner. When detecting caspase-3 activity, approximately 75.4% cells presented proteases activities at 6 h post-PDT. Caspase family proteases were activated comparatively slightly and slowly after PDT.
Compared with the control groups, in the ZnPc-PDT groups the ratio of cytosol cytochrome c (15 kDa) to β-actin (43 kDa) assessed by Western blot analysis increased significantly (P<0.01). Significant difference existed between 6 h and 12 h post-PDT (P<0.01). However the difference between 12 h and 24 h points was minor (P>0.05) (Figure 5A). Immunochemistry assay showed that after ZnPc-PDT, cytochrome c translocated from its original location adjacent to nucleus and dispersed into cytosol and the positive particles were stained obviously darker (Figure 5B). The 2 indexes showed that ZnPc-PDT induced evident release of cytochrome c from the mitochondria into the cytosol.
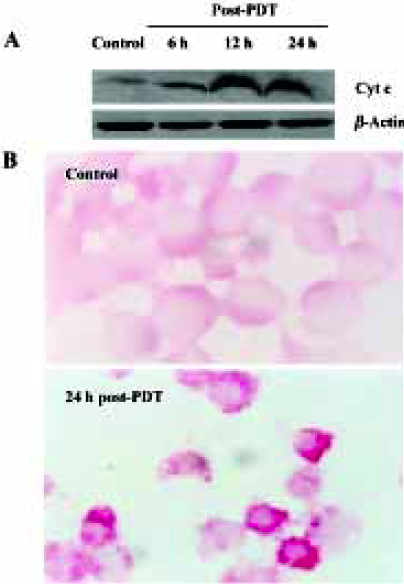
Summing up the results from different observations as previously discussed, we concluded that ZnPc-PDT could induce HL60 cells apoptosis through mitochondria-dependent ways.
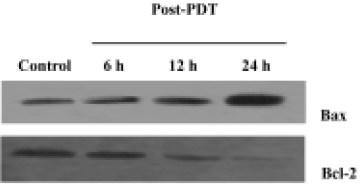
Effects of ZnPc-PDT on the expression of Bcl-2 and Bax Western blot assays showed that ZnPc-PDT could down-regulate Bcl-2 expression and up-regulate Bax expression. After PDT, the ratio of Bax to Bcl-2 increased significantly in a time-dependent manner (Figure 6).
Discussion
PDT has emerged as a promising therapeutic procedure for the treatment of many tumors. The cytotoxic efficacy relies on a bimodal protocol comprised of chemical-photosensitizer and light-irradiation. Separately, neither the photosensitizer nor the light is toxic[1,11]. The mechanism of the PDT-mediated cytotoxic effects is not well defined. However it is generally believed that the apoptotic response of PDT is critical for its therapeutic efficacy and the generation of ROS is essential for PDT effects[2,3,25]. Some results have demonstrated that PDT could induce the permeability transition pore open and the opening leads to the loss of integrity of the outer mitochondrial membrane, thus releasing the inter-membrane proteins, such as cytochrome c, into the cytosol[12–16]. When released from the mitochondria, cytochrome c combines with an inactive protease precursor, procaspase-9, to form the “apoptosome”. The apoptosome attacks procaspase-3 and cleaves it to form active caspase-3, which can lead to DNA breakage, nuclear chromatin condensation, then cause cell death[20,21].
A number of studies have documented that cell death response to PDT varies due to the physical properties of the photosensitizer employed, PDT dose and the cell type[4–8]. Different subcellular localization of the photosensitizer results in different responses[6,7]. Even the same photosensitizer-mediated PDT could lead to different outcomes. At low doses, the cellular machinery for apoptosis is activated, however, higher doses lead to the inhibition of apoptosis with cell death via a necrotic process[4,8]. In the present study, a novel photosensitizer agent, ZnPcS2P2, developed in China, was employed. Our previous studies have demonstrated that ZnPc-PDT could exert significant photodamage to HL60 cells[11]. In the present study, 0.5 µmol/L drug level was selected as the experimental condition. Under the condition, ZnPc-PDT could inhibit the proliferation of HL60 cells at 51%.
It is well known that there are two major pathways that are involved in the initiation of apoptosis: the “extrinsic” death receptor pathway and the “intrinsic” mitochondrial pathway. The present study provides direct evidence for the role of mitochondria in HL60 cells apoptosis induced by ZnPc-PDT. Cytochrome c appeared in the cytosol 6 h following light activation of ZnPcS2P2. The main cytochrome c release occurred later, between 6 h and 12 h after PDT. However, caspase-3 could be quickly activated by ZnPc-PDT. Remarkable activity of caspase-3 was observed 6 h post-PDT in almost all cells. According to recent studies[28], during genotoxic stress-induced apoptosis, the release of cytochrome c from mitochondria occurrs in 2 distinct stages. The early release of low levels of cytochrome c into the cytosol precedes the activation of caspase 9 and 3. The late stage cytochrome c release results in a drastic loss of mitochondrial cytochrome c and the activities of caspases contribute to the late cytochrome c release mentioned earlier. In the present study, PDT induced the early release of low levels of cytochrome c into the cytosol in the first 6 h. The cytochrome c then combined with procaspase-9 to form the “apoptosome”. The apoptosome activated caspase-3 that induced the late cytochrome c release. Therefore, between 6 h and 12 h post-PDT, the main cytochrome c release occurred. At the time of cytochrome c release and remarkable caspase-3 activation (6 h after PDT), only a 12.6% fraction of the cells detected disruption of Δψm. The data are in agreement with the previous conclusion[19] that the dissipation of Δψm is not required for the release of cytochrome c from the mitochondria. Also, the up-regulation of pro-apoptotic protein Bax, and the down-regulation of anti-apoptotic protein Bcl-2 by ZnPc-PDT appeared significantly and could result in activating mitochondria-mediated apoptosis. The results coincide with those of other previously published studies[25,29]. It is also reported that in many cell types, there is delicate coordination and cross talk between the extrinsic and intrinsic pathways that leads to the activation of the executioner caspase cascade[30,31]. Therefore, the study of the role of the death receptor pathway in the apoptosis induced by ZnPc-PDT should be pursued.
Thus, we conclude that the main mechanism for the apoptosis in HL60 cells induced by ZnPc-PDT appeared to be mitochondria-mediated pathway accompanied with cytochrome c release from the mitochondria into the cytosol followed by the activation of caspase-3. Our findings would provide a fundamental basis for the clinical application of ZnPc-PDT.
Acknowledgments
We wish to thank Prof Jin-ling HUANG and Prof Nai-sheng CHEN for the gift of ZnPcS2P2, Prof Shu-sen XIE and Prof Rong CHEN for instructions in semi-conductor laser operation.
References
- Moan J, Berg K. Photochemotherapy of cancer: experimental research. Photochem Photobiol 1992;55:931-48.
- Henderson B, Dougherty TJ. How does photodynamic therapy work? Photochem Photobiol 1992;55:145-57.
- Niedre M, Patterson MS, Wilson BC. Direct near-infrared luminescence detection of singlet oxygen generated by photodynamic therapy in cells in vitro and tissues in vivo. Photochem Photobiol 2002;75:382-91.
- Wyld L, Reed MW, Brown NJ. Differential cell death response to photodynamic therapy is dependent on dose and cell type. Br J Cancer 2001;84:1384-6.
- Luo Y, Kessel D. Initiation of apoptosis versus necrosis by photodynamic therapy with chloroaluminum phthalocyanine. Photochem Photobiol 1997;66:479-83.
- Kessel D, Luo Y, Deng Y. The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem Photobiol 1997;65:422-6.
- Noodt BB, Berg K, Stokke T, Pen Q, Nosland JM. Different apoptotic pathways are induced from various intra cellular sites by tetraphenylporphyrins and light. Br J Cancer 1999;79:72-81.
- Kessel D, Poretz RD. Sites of photodamage induced by photodynamic therapy with a chlorine 6 triacetoxymethyl ester (CAME). Photochem Photobiol 2000;71:94-6.
- Huang JL, Chen NS, Huang JD, Liu ES, Xue JP, Yang SL, et al. Synthesis, peculiarity and anti-tumor potency of the double affinity photosensitizer-zinc phthalocyanine. Sci China 2000;30:481-8. (Series B) (Chin).
- Huang JL, Chen NS, Huang JD, Liu ES, Xue JP, Yang SL, et al. Metal phthalocyanine as photosensitizer for photodynamic therapy (PDT). Sci China 2001;44:113-22. (Series B).
- Huang HF, Chen YZ, Wu Y. Experimental studies of the effects of ZnPcS2P2-based-photodynamic therapy on bone marrow purging. Natl Chin Med J 2005;118:105-10.
- Vantieghem A, Xu Y, Declercq W, Vandenabeele P, Denecker G, Vandenheede JR, et al. Different pathways mediate cytochrome C release after photodynamic therapy with hypericin. Photo-chem Photobiol 2001;74:1333-42.
- Matroule JY, Carthy CM, Granville DJ, Jolois O, Hunt DW, Piette J. Mechanism of colon cancer cell apoptosis mediated by pyropheophorbide-a methylester photosensitization. Oncogene 2001;20:4070-84.
- Casas A, Fukuda H, Di Venosa G, Batle A. Photosensitization and mechanism of cytotoxicity induced by the use of ALA derivatives in photodynamic therapy. Br J Cancer 2001;85:279-84.
- Chiu SM, Evans H, Lam M, Nieminen A, Oleinick NL. Phthalocyanine 4 photodynamic therapy-induced apoptosis of mouse L5178Y-R cells results from a delayed but extensive release of cytochrome C from mitochondria. Cancer Lett 2001;165:51-8.
- Kessel D, Luo Y. Mitochondrial photodamage and PDT-induced apoptosis. J Photochem Photobiol B 1998;42:89-95.
- Morgan J, Oseroff AR. Mitochondria-based photodynamic anti-cancer therapy. Adv Drug Deliv Rev 2001;49:71-86.
- Belichenko I, Morishima N, Separovic D. Caspase-resistant Vimentin suppresses apoptosis after photodynamic treatment with a silicon phthalocyanine in Jurkat cells. Arch Biochem Biophys 2001;390:57-63.
- Chiu SM, Oleinick NL. Dissociation of mitochondrial depolarization from cytochrome C release during apoptosis induced by photodynamic therapy. Br J Cancer 2001;84:1099-106.
- Gad F, Viau G, Boushira M, Bertrand R, Bissonnette R. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg 2001;5:8-13.
- Xue LY, Chiu SM, Oleonick NL. Photodynamic therapy-induced death of MCF-7 human breast cancer cells: a role for caspase-3 in the late steps of apoptosis but not for the clinical lethal event. Exp Cell Res 2001;263:145-55.
- He J, Whitacre CM, Xue LY, Berger NA, Oleinick NL. Protease activation and cleavage of poly (ADP-ribose) polymerase: an integral part of apoptosis in response to photodynamic treatment. Cancer Res 1998;58:940-6.
- Reed JC. Double identity for proteins of Bcl-2 family. Nature 1997;387:773-6.
- Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S, et al. Bcl-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem 1996;60:23-32.
- Xue LY, Chiu SM, Oleinick NL. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc4. Oncogene 2001;20:3420-7.
- Wu Y, Chen YZ, Huang HF, Chen P. Recombinant fibronectin polypeptide antagonizes hepatic failure induced by endotixin in mice. Acta Pharmacol Sin 2004;25:283-8.
- Yang J, Liu XS, Kapil B, Caryn NK, Ana MI, Cai JY, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997;275:1129-32.
- Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ 2000;7:227-33.
- Srivastava M, Ahmad N, Gupta S, Mukhtar H. Involvement of Bcl-2 and Bax in photodynamic therapy-mediated apoptosis. Antisense Bcl-2 oligonucleotide sensitizes RIF 1 cells to photodynamic therapy apoptosis. J Biol Chem 2001;276:15481-8.
- Jung JY, Kim WJ. Involvement of mitochondrial- and Fas-mediated dual mechanism in CoCl2-induced apoptosis of rat PC12 cells. Neurosci Lett 2004;371:85-90.
- Khosravi-Far R, Esposti MD. Death receptor signals to mitochondria. Cancer Biol Ther 2004;3:1051-7.