
Protective effect of esculetin against oxidative stress-induced cell damage via scavenging reactive oxygen species
Introduction
A redox imbalance in a healthy living system leads to the malfunctioning of cells, which ultimately results in various reactive oxygen species (ROS) related diseases, including cancer, neurological degeneration, arthritis, and the aging process[1,2]. The consequences of this redox imbalance become even more harmful when genetic variation impairs the normal degradation of these altered proteins. Therefore, therapeutic strategies should be focused on the reduction of free radical formation and scavenging of free radicals[3].
Plants, including fruits, vegetables, and medicinal herbs, may contain a wide variety of free radical scavenging molecules such as polyphenols. Natural polyphenols can be divided into several different classes depending on their basic chemical structure ranging from simple molecules to highly polymerized compounds. Coumarin, which is known as 1,2-benzopyrone, consists of fused benzene and α-pyrone rings, and is an important low-molecular weight phenolic group[4] widely used for the prevention and treatment of venous thromboembolism, myocardial infarction, and stroke[5]. Natural and synthetic coumarins possess anti-oxidant, anti-inflammatory, anticoagulation, and anticancer activities[6]. For the in vivo experiment, rats treated with coumarin ameliorated the hepatotoxicity induced by oxidative stress[7]. Therefore, researchers are looking for natural anti-oxidants with strong pharmacological action and less cytotoxic properties. The efficiency of phenolic compounds as antiradicals and anti-oxidants is variable and depends on many factors such as the number of hydroxyl groups bonded to the aromatic ring, the site of bonding, and the mutual position of hydroxyls in the aromatic ring. For example, previous studies have found that the advantageous effect of an o-dihydroxy substitution in the aromatic ring of phenol for radical scavenging activity is achieved by producing quinone via hydrogen donation[8,9].
Here, we demonstrate that esculetin (6,7-dihydroxycoumarin) was found to possess cytoprotective properties against oxidative stress-induced cell damage.
Materials and methods
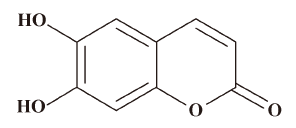
Cell culture and reagents Chinese hamster lung fibroblast (V79-4) cells were obtained from the American type culture collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal calf serum (FCS), streptomycin (100 µg/mL) and penicillin (100 U/mL) at 37 °C in a humidified atmosphere of 5% CO2. Esculetin (Figure 1) was purchased from Wako Pure Chemicals (Tokyo, Japan) and dissolved in dimethylsulfoxide (DMSO) and the final concentration of DMSO did not exceed 0.02%. The 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), and 2’,7’-dichlorodihydrofluorescein diacetate (DCF-DA) were purchased from Sigma Chemical Company (St Louis, MO, USA). The thiobarbituric acid was purchased from BDH laboratories (Poole, Dorset, UK). Anti-phospho histone H2A.X antibody was purchased from Upstate Biotechnology (Lake Placid, NY, USA). All other chemicals and reagents used were of analytical grade.
DPPH radical scavenging activity Esculetin at 0.1, 1, and 10 µg/mL was added to a 1×10–4 mol/L solution of DPPH in methanol, and the reaction mixture was shaken vigorously. After 5 h, the remaining DPPH was determined by measuring its absorbance at 520 nm[10]. The DPPH radical scavenging activity (%) was calculated as; (optical density of DPPH radical treatment-optical density of esculetin with DPPH radical treatment)/(optical density of DPPH radical treatment)×100.
Detection of hydroxyl radical The hydroxyl radicals were generated by the Fenton reaction (H2O2+FeSO4), and then reacted with a nitrone spin trap, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). The resultant DMPO-OH adducts were detected using an electron spin resonance (ESR) spectrometer. The ESR spectrum was recorded using a JES-FA ESR spectrometer (JEOL, Tokyo, Japan), at 2.5 min after mixing phosphate buffer solution (pH 7.4) with 0.2 mL of 0.3 mol/L DMPO, 0.2 mL of 10 mmol/L FeSO4, 0.2 mL of 10 mmol/L H2O2, and esculetin at 10 µg/mL. The parameters of the ESR spectrometer were set at a magnetic field of 336.5 mT, power of 1.00 mW, frequency of 9.4380 GHz, modulation amplitude of 0.2 mT, gain of 200, scan time of 0.5 min, scan width of 10 mT, time constant of 0.03 s, and a temperature of 25 °C[11,12].
Intracellular ROS measurement The DCF-DA method was used to detect the levels of intracellular ROS[13]. The V79-4 cells were seeded in a 96-well plate at 2×104 cells/well. 16 h after plating, the cells were treated with esculetin at 0.1, 1, and 10 µg/mL. After 30 min, 1 mmol/L of H2O2 was added to the plate. The cells were incubated for an additional 30 min at 37 °C. After adding 25 µmol/L of DCF-DA solution for 10 min, the fluorescence of 2’,7’-dichlorofluorescein was detected using a Perkin Elmer LS-5B spectrofluorometer and using flow cytometry (Becton Dickinson, Mountain View, CA, USA), respectively. The intracellular ROS scavenging activity (%) was calculated as follows; (optical density of H2O2 treatment–optical density of esculetin with H2O2 treatment)/(optical density of H2O2 treatment)×100. The image analysis for the generation of intracellular ROS was carried out by seeding the cells on a cover-slip loaded 6 well plate at 2×105 cells/well. 16 h after plating, the cells were treated with 10 µg/mL of esculetin. After 30 min, 1 mmol/L of H2O2 was added to the plate. After changing the media, 100 µmol/L of DCF-DA was added to each well and was incubated for an additional 30 min at 37 °C. After washing with phosphate-buffered saline (PBS), the stained cells were mounted onto microscope slides in mounting medium (DAKO, Carpinteria, CA, USA). The microscopic images were collected using the Laser Scanning Microscope 5 PASCAL program (Carl Zeiss, Jena, Germany) on a confocal microscope.
Lipid peroxidation detection Lipid peroxidation was assayed by the measurement of related substances that react with thiobarbituric acid (TBARS)[14]. The V79-4 cells were seeded in a culture dish at a concentration of 1×105 cells/mL, and treated with esculetin at 10 µg/mL after 16 h plating. And then H2O2 1 mmol/L was added to the plate After 1 h, which was incubated for a further 24 h. The cells were then washed with cold PBS, scraped and homogenized in ice-cold 1.15% KCl. About 100 µL of cell lysates was combined with 0.2 mL of 8.1% SDS, 1.5 mL of 20% acetic acid (adjusted to pH 3.5) and 1.5 mL of 0.8% thiobarbituric acid (TBA). The mixture was adjusted to a final volume of 4 mL with distilled water and heated to 95 °C for 2 h. After cooling to room temperature, 5 mL of a mixture of n-butanol and pyridine (15:1) was added to each sample, and the mixture was shaken vigorously. After centrifugation at 1000×g for 10 min, the supernatant fraction was isolated, and the absorbance was measured at 532 nm.
Protein carbonyl formation The cells were treated with 10 µg/mL of esculetin, and after 1 h, 1 mmol/L of H2O2 was added to the plate, and the mixture was incubated for 24 h. The amount of carbonyl formation in protein was determined using an Oxiselect protein carbonyl ELISA kit purchased from Cell Biolabs (San Diego, CA, USA) according to the manufacturer’s instructions. Cellular protein was isolated using protein lysis buffer (50 mmol/L Tris (pH 7.5), 10 mmol/L EDTA (pH 8), 1 mmol/L phenylmethanesulfonyl fluoride (PMSF) and quantified using a spectrophotometer.
Western blot analysis The cells were treated with 10 µg/mL of esculetin, and after 1 h, 1 mmol/L of H2O2 was added to the plate, and the mixture was incubated for 24 h. The cells were harvested, and washed twice with PBS. The harvested cells were then lysed on ice for 30 min in 100 µL of lysis buffer (120 mmol/L NaCl, 40 mmol/L Tris [pH 8], 0.1% NP 40) and centrifuged at 13 000×g for 15 min. Supernatants were collected from the lysates and protein concentrations were determined. Aliquots of the lysates (40 µg of protein) were boiled for 5 min and electrophoresed in 10% SDS-polyacrylamide gel. Blots in the gels were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), which were then incubated with primary antibody. The membranes were further incubated with secondary immunoglobulin G (IgG)-horseradish peroxidase conjugates (Pierce, Rockland, IL, USA), and then exposed to X-ray film. The protein bands were detected using an enhanced chemiluminescence Western blotting detection kit (Amersham, Little Chalfont, Buckinghamshire, UK).
Immunocytochemistry Cells plated on coverslips were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 in PBS for 2.5 min. Cells were treated with blocking medium (3% bovine serum albumin in PBS) for 1 h and incubated with anti-phospho histone H2A.X antibody diluted in blocking medium for 2 h. Primary phospho histone H2A.X antibody was detected by a 1:500 dilution of fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Jackson Immuno Research Laboratories, West Glove, PA, USA) for 1 h. After washing with PBS, the stained cells were mounted onto microscope slides in mounting medium with 4’,6’-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector, Burlingame, CA, USA). The images were collected using the Laser Scanning Microscope 5 PASCAL program (Carl Zeiss, Jena, Germany) on a confocal microscope.
Cell viability The effect of esculetin on the viability of cells was determined by the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which is based on the cleavage of a tetrazolium salt by mitochondrial dehydrogenase in viable cells[15]. The cells were seeded in a 96 well plate at a concentration of 1×105 cells/mL. At 16 h after plating, the cells were treated with 10 µg/mL of esculetin, and 1 h later 1 mmol/L H2O2 was added to the plate and incubated for an additional 24 h at 37 °C. 50 µL of MTT stock solution (2 mg/mL) was then added to each well with a total reaction volume of 200 µL. After incubation for 4 h, the plate was centrifuged at 800×g for 5 min and the supernatants aspirated. The formazan crystals in each well were dissolved in 150 µL DMSO and the A540 was read on a scanning multi-well spectrophotometer.
Statistical analysis The data are presented as the means±SEM. The results were subjected to ANOVA using the Tukey test to analyze the difference. P<0.05 was considered significantly.
Results
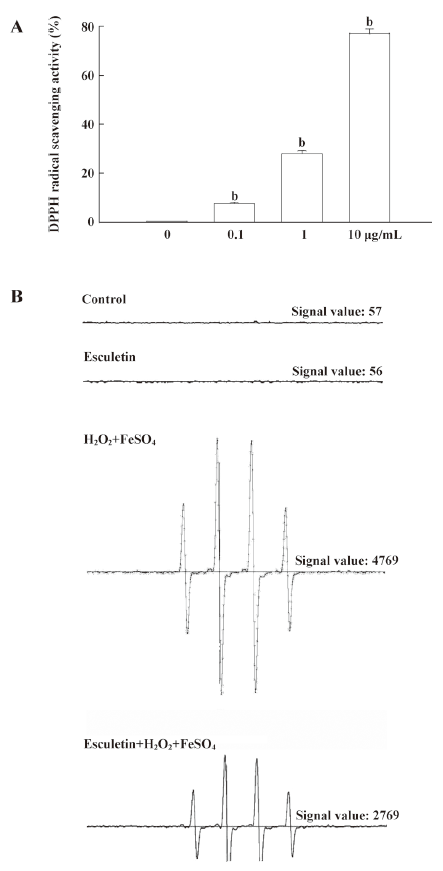
Radical scavenging activity of esculetin in a cell-free system The radical scavenging effects of esculetin on DPPH radicals and hydroxyl radicals were measured. The DPPH radical scavenging activity of esculetin was 8% at 0.1 µg/mL, 28% at 1 µg/mL, and 77% at 10 µg/mL (Figure 2A). In addition, the hydroxyl radicals generated by the Fenton reaction (FeSO4+H2O2) in a cell-free system were detected by ESR spectrometry. The ESR results revealed that a specific signal was not clearly detected in the control and the 10 µg/mL of esculetin; however, the signal of the hydroxyl radical increased up to 4769 in the FeSO4+H2O2 system. Esculetin treatment was found to decrease the hydroxyl radical signal to 2769 (Figure 2B).
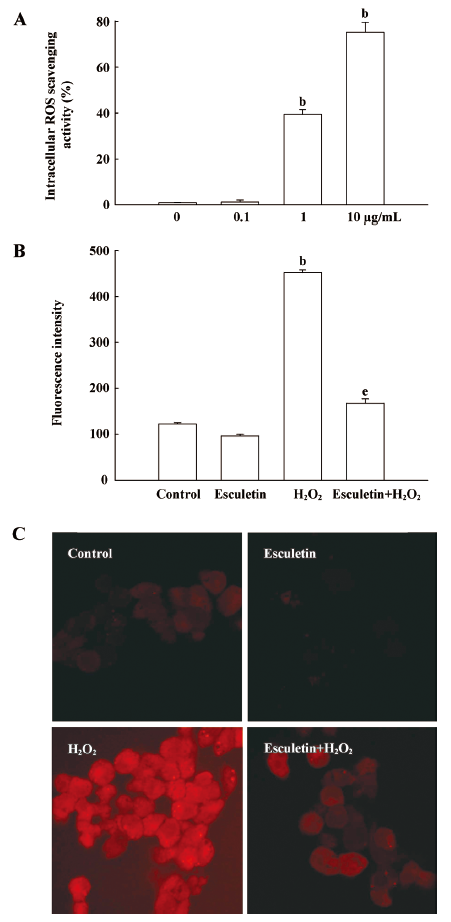
Intracellular ROS scavenging activity of esculetin The intracellular ROS scavenging activity of esculetin in V79-4 cells after H2O2 treatment was detected by the DCF-DA assay. The fluorescence spectrometric data revealed that the intracellular ROS scavenging activity of esculetin was 1% at 0.1 µg/mL, 40% at 1 µg/mL, and 75% at 10 µg/mL (Figure 3A). Moreover, the fluorescence intensity of DCF-DA staining was measured using a flow cytometer and a confocal microscope. The level of ROS was detected using a flow cytometer, which revealed a fluorescence intensity value of 167 for the ROS stained by DCF-DA fluorescence dye in H2O2 treated cells with 10 µg/mL of esculetin, compared with a fluorescence intensity value of 452 in the H2O2 treated cells (Figure 3B). Moreover, the results of confocal microscopy revealed that esculetin reduced the red fluorescence intensity induced by H2O2 treatment (Figure 3C), thus indicating a reduction in ROS generation. Taken together, these results suggest that esculetin possesses an anti-oxidant effect via a radical scavenging effect.
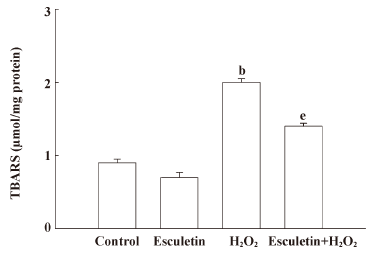
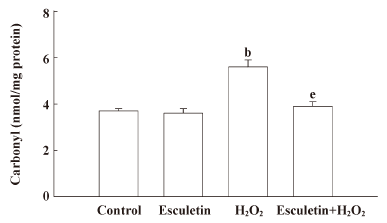
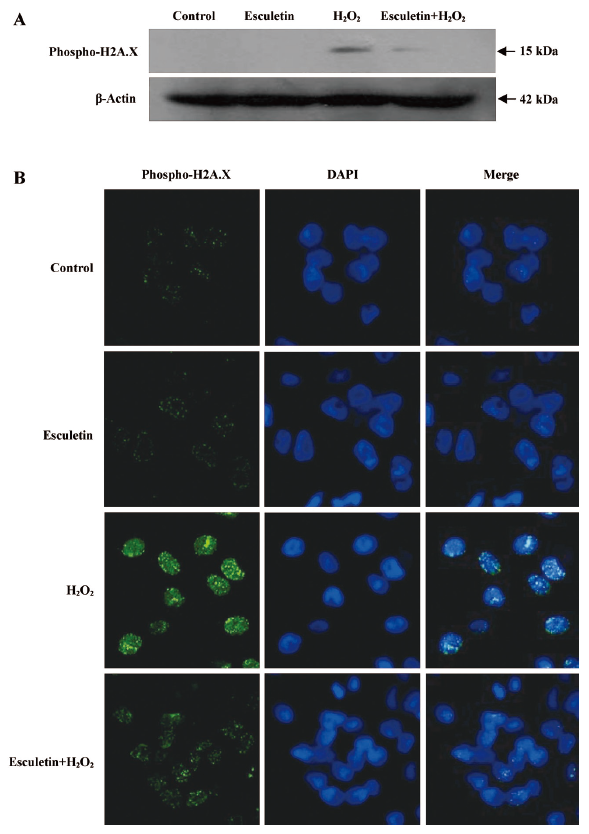
Effect of esculetin against the damage of cellular components induced by H2O2 treatment The H2O2-induced damage of cellular components represents the primary cause of cell viability loss. The effect of esculetin on the damage of membrane lipid, protein, and cellular DNA in H2O2-treated cells was investigated. V79-4 cells exposed to H2O2 had increased lipid peroxidation, as evidenced by the generation of TBARS. However, esculetin treatment was found to prevent the H2O2-induced peroxidation of lipids (Figure 4). The protein carbonyl formation serves as a biomarker for cellular oxidative protein damage[16]. Moreover, the accumulation of oxidatively modified protein carbonyls may disrupt cellular function either by the loss of catalytic and structural integrity or by the interruption of regulatory pathways[17]. Moreover, the protein carbonyl content in cells increased significantly after H2O2 treatment, whereas esculetin prevented the H2O2-induced protein carbonyl formation (Figure 5). Further, damage to cellular DNA induced by H2O2 exposure was detected by phospho histone-H2A.X expression in nucleus. The phosphorylation of the nuclear histone H2A.X, which is a sensitive marker for breaks of double stranded DNA[18], increased in the H2O2-treated cells, as shown by Western blot and immuno-fluorescence imaging (Figure 6A,B). However, esculetin treatment in H2O2-treated cells decreased the expression of phospho H2A.X. These results suggest that esculetin inhibits the damage of cellular components induced by H2O2.
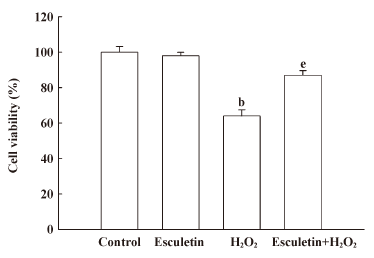
Protective effect of esculetin on cell damage induced by H2O2 The protective effect of esculetin on the cell survival in H2O2-treated V79-4 cells was also assessed. The cells were treated with 10 µg/mL of esculetin for 1 h prior to the addition of H2O2. The cell viability was determined 24 h later by MTT assay (Figure 7). Treatment with esculetin at 10 µg/mL in V79-4 cells did not show cytotoxicity, compared with the control. Combination with esculetin at 10 µg/mL and H2O2 increased 87% in the cell survival rate, compared with 64% in H2O2 treated cells. The findings suggest that esculetin results in the protection of cell viability by inhibiting the damage of the cellular components induced by H2O2.
Discussion
Coumarins are lactones of cis-o-hydroxycinnamic acid derivatives, which belong to the phenol compounds with a basic skeleton of C6+C3[19]. Also, coumarins are a diverse group of natural compounds essentially found in green plants. As substitutions can occur at any of the 6 available sites of their basic molecular moiety (1,2-benzopyrone), these compounds are variable in structure. This structural diversity results in the multiple biological properties of coumarin that help reducing the risk of human disease. In a recent paper[20], coumarin itself and 7-hydroxycoumarin showed evidence of in vitro antiproliferative and in vivo antitumor activities. Moreover, coumarins possess anti-oxidant activities, probably due to their structural analogy with flavonoids and benzophenones[21]. For example, esculetin isolated from the cortex of Fraxius chinensis showed anti-photoaging activity through anti-oxidant activities[22]. Coumarins in plants are present in the free form or glycosides, and are emerging as potent therapeutic drugs for free radical-mediated diseases. Protection of the cellular environment from oxidative stress is dependent on the chemical structure of anti-oxidants, suggesting that the radical-scavenging effects of coumarins are correlated with the number and position of the hydroxyl groups. A similar correlation was also reported for flavonoids[23] and cinnamic acid[24]. In general, the radical scavenging activity of flavonoids depends on the molecular structure and the substitution pattern of the hydroxyl groups, on the availability of phenolic hydrogens and on the possibility of stabilization of the resulting phenoxyl radicals via hydrogen bonding or by expanded electron delocalization[25]. A previous report has also found that the phenolic hydroxyl groups, which have o-dihydroxy group (catechol structure), enhance and play an important role in antiradical efficacy of simple coumarins[26]. These results are consistent with a previous report that the radical scavenging activity was higher for caffeic acid bearing o-dihydroxy group than methoxy-substituted derivatives[27]. Thus it can be inferred that the resonance structures of the radical derived from esculetin is especially stable as a result of the o-quinone form of the resonance structure. A recent paper showed consistent results that coumarin derivatives from the dihydroxy group were more potent DPPH radical scavengers than monohydroxy, free hydroxyl, methoxyl, dihydro, and coumarin derivatives[6]. Therefore, our study demonstrated that esculetin possessed anti-oxidant properties and provided cytoprotection against oxidative stress. Esculetin showed evidence of direct radical quenching as shown by the DPPH radical and hydroxyl radical and it might be related with catechol structure of esculetin. In addition, esculetin showed intracellular ROS scavenging activities, suggesting that it might be involved in activation of anti-oxidant enzymes. Oxidative stress occurs when there is an imbalance between pro-oxidant processes and the anti-oxidant defense system in favor of the former, and will ultimately lead to a free radical attack of the lipid, protein, and DNA. H2O2 is a major component of the ROS produced intracellularly and a cause of oxidative damage[28]. Lipid peroxidation is one of the major mechanisms of cellular injury caused by H2O2[29]. In our study, cells exposed to H2O2 showed an increase in lipid peroxidation. However, esculetin was found to decrease the H2O2-induced lipid peroxidation, indicating the cytoprotective properties of esculetin against H2O2 induced lipid damage. Moreover, the oxidative damage of the amino acid residues of the proteins resulted in the formation of carbonyl derivatives and can seriously compromise cellular integrity[30]. The protein carbonyl content in cells increased significantly after H2O2 treatment, whereas esculetin treatment was found to prevent the H2O2-induced protein carbonyl formation. In addition, H2O2 is a genotoxic agent and is known to induce oxidative DNA damage[31]. In this study, H2O2-induced DNA damage was assessed using the expression of the phospho histone H2A.X to detect DNA strand breakage. The exposure of cells to H2O2 increased phosphorylation of the nuclear histone H2A.X. However, esculetin treatment in H2O2-treated cells decreased the expression of the nuclear histone H2A.X, thus indicating the protective effect of esculetin against H2O2-induced DNA damage. Therefore, esculetin provides cytoprotection against H2O2-induced lipid peroxidation, protein carbonyl, and DNA damage by scavenging intracellular H2O2.
In summary, many biochemical and clinical studies suggest that natural and synthetic anti-oxidant compounds help treat diseases mediated by oxidative stress[32]. The results presented in this report indicate that esculetin efficiently attenuated oxidative stress-induced cell damage via its anti-oxidant properties. Thus, esculetin might be useful in the development of functional food and raw materials of medicine.
Acknowledgements
This research was carried out under the program of the Basic Atomic Energy Research Institute (BAERI) which is a part of the Nuclear R&D Programs and in part from the study of the DNA repair regulation with the disease program [M1063901] funded by the Ministry of Science & Technology of Korea (KOSEF).
References
- Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr 1996;16:33-50.
- Willcox JK, Ash SL, Catignani GL. Antioxidants and prevention of chronic disease. Crit Rev Food Sci Nutr 2004;44:275-95.
- Berg D, Youdim MB, Riederer P. Redox imbalance. Cell Tissue Res 2004;318:201-13.
- Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Nicolaides DN. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr Pharm Des 2004;10:3813-33.
- Voora D, McLeod HL, Eby C, Gage BF. The pharmacogenetics of coumarin therapy. Pharmacogenomics 2005;6:503-13.
- Lin HC, Tsai SH, Chen CS, Chang YC, Lee CM, Lai ZY, et al. Structure-activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Biochem Pharmacol 2008;75:1416-25.
- Lin WL, Wang CJ, Tsai YY, Liu CL, Hwang JM, Tseng TH. Inhibitory effect of esculetin on oxidative damage induced by t-butyl hydroperoxide in rat liver. Arch Toxicol 2000;74:467-72.
- Joyeux M, Lobstein A, Anton R, Mortier F. Comparative antilipoperoxidant, antinecrotic and scavenging properties of terpenes and biflavones from Ginkgo and some flavonoids. Planta Med 1995;61:126-9.
- Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med 1996;20:933-56.
- Lo SF, Nalawade SM, Mulabagal V, Matthew S, Chen CL, Kuo CL, et al. In vitro propagation by asymbiotic seed germination and 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity studies of tissue culture raised plants of three medicinally important species of Dendrobium. Biol Pharm Bull 2004;27:731-5.
- Yoshimura Y, Inomata T, Nakazawa H, Kubo H, Yamaguchi F, Ariga T. Evaluation of free radical scavenging activities of antioxidants with an H2O2/NaOH/DMSO system by electron spin resonance. J Agric Food Chem 1999;47:4653-6.
- Rimbach G, Weinberg PD, de Pascual-Teresa S, Alonso MG, Ewins BA, Turner R, . Sulfation of genistein alters its antioxidant properties and its effect on platelet aggregation and monocyte and endothelial function. Biochim Biophys Acta 2004; 1670: 229–37.
- Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, Chen W, Knapp W, Zlabinger GJ. A microplate assay for the detection of oxidative products using 2’,7’-dichlorofluorescin-diacetate. J Immunol Meth 1992;156:39-45.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351-8.
- Carmichael J, DeGraff WG, Gazdar AF. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 1987;47:936-41.
- Szweda LI, Stadtman ER. Oxidative modification of glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides by an iron(II)-citrate complex. Arch Biochem Biophys 1993;301:391-5.
- Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci 2000;899:191-208.
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998;273:5858-68.
- Cai YZ, Mei S, Jie X, Luo Q, Corke H. Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci 2006;78:2872-88.
- Kostova I. Synthetic and natural coumarins as cytotoxic agents. Curr Med Chem Anticancer Agents 2005;5:29-46.
- Farombi EO, Nwaokeafor IA. Anti-oxidant mechanisms of kolaviron: studies on serum lipoprotein oxidation, metal chelation and oxidative membrane damage in rats. Clin Exp Pharmacol Physiol 2005;32:667-74.
- Lee BC, Lee SY, Lee HJ, Sim GS, Kim JH, Kim JH, et al. Anti-oxidative and photo-protective effects of coumarins isolated from Fraxinus chinensis. Arch Pharm Res 2007;30:1293-301.
- Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J Nutr Biochem 2002;13:572-84.
- Siquet C, Paiva-Martins F, Lima JL, Reis S, Borges F. Antioxidant profile of dihydroxy- and trihydroxyphenolic acids-a structure-activity relationship study. Free Radic Res 2006;40:433-42.
- Amic D, Davidovic-Amic D, Beslo D, Trinajstic N. Structure-radical scavenging activity relationships of flavonoids. Croat Chem Acta 2003;76:55-61.
- Wu CR, Huang MY, Lin YT, Ju HY, Ching H. Antioxidant properties of Cortex Fraxini and its simple coumarins. Food Chem 2007;104:1464-71.
- Chang YC, Lee FW, Chen CS, Huang ST, Tsai SH, Huang SH, et al. Structure-activity relationship of C6-C3 phenylpropanoids on xanthine oxidase-inhibiting and free radical-scavenging activities. Free Radic Biol Med 2007;43:1541-51.
- Krohn K, Maier J, Paschke R. Mechanisms of disease: hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat Clin Pract Endocrinol Metab 2007;3:713-20.
- Young J, McKinney SB, Ross BM, Wahle KW, Boyle SP. Biomarkers of oxidative stress in schizophrenic and control subjects. Prostaglandins Leukot Essent Fatty Acids 2007;76:73-85.
- Hawkins CL, Davies MJ. Generation and propagation of radical reactions on proteins. Biochim Biophys Acta 2001;1504:196-219.
- Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett 1991;281:9-19.
- Lee JC, Lim KT, Jang YS. Identification of Rhus verniciflua stokes compounds that exhibit free radical scavenging and anti-apoptotic properties. Biochim Biophys Acta 2002;1570:181-91.