
Uric acid stimulates endothelin-1 gene expression associated with NADPH oxidase in human aortic smooth muscle cells
Introduction
Uric acid is an intermediate product of the purine degradation pathway in the cell. An elevated serum uric acid in humans is associated with hypertension[1] and cardiovascular diseases[2]. All of these conditions are thought to be mediated by oxidative stress[3]. Experimental studies have shown that hyperuricemic rats (induced by blocking uricase) also develop hypertension, and vascular diseases[4]. Physiological concentrations of soluble microcrystal-free uric acid induce gene expression of chemokines and growth factors, such as monocyte chemoattractant protein (MCP)-1 and platelet-derived growth factor[5,6], and stimulate proliferation of vascular smooth muscle cells with the activation of mitogen-activated protein kinases (MAPK)[6,7]. The effects of uric acid may involve complex but poorly understood redox-dependent pathways. Uric acid-induced MCP-1 expression in vascular smooth muscle cells was attenuated by antioxidants, suggesting the involvement of a redox-dependent mechanism[5]. More recently, it has been shown that soluble uric acid stimulated an increase in NADPH oxidase activity and reactive oxygen species (ROS) production in mature adipocytes[8]. What remains unclear is whether ROS also play a role in the direct effects of uric acid on human aortic smooth muscle cells (HASMC).
Endothelin-1 (ET-1) is a potent vasoconstrictor derived from the endothelium[9]. Patients with moderate to severe hypertension reveal increased vascular levels of prepro-ET-1 mRNA[9]. Elevated levels of ET-1 have emerged as one of the most important predictors of myocardial infarction, stroke, and vascular death[10]. It has been shown that the plasma levels of ET-1 were significantly associated with uric acid by univariate analysis in an epidemiological study[11]. We previously reported that ROS via the activation of extracellular signal-regulated kinase (ERK) and the nuclear transcription factor activator protein-1 (AP-1) are essential for cell proliferation and ET-1 gene expression in rat aortic smooth muscle cells[12]. However, the effect of uric acid on cell proliferation and ET-1 gene expression in HASMC remains to be elucidated.
Despite the striking common features of uric acid and ET-1 in the development of hypertension and cardiovascular diseases, there are no data on whether uric acid-induced vascular changes in blood vessels are also related to ET-1 expression. In this study we attempted to clarify the relationship of uric acid with ET-1 expression, and investigated the effect of uric acid on ET-1 synthesis in human vascular cells.
Materials and methods
Materials Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum, and tissue culture reagents were from Life Technologies, Inc (Gaithersburg, MD, USA). An ET-1 cDNA probe was obtained as previously described[13]. Wild type (204 bp) or AP-1 mutant of ET-1 promoter was fused with the chloramphenicol acetyltransferase (CAT) reporter gene (ET-1 promoter-CAT plasmid); PBLCAT2 (containing CAT reporter gene with its promoter) and PBLCAT3 (containing CAT gene only) were constructed as previously described[13]. 2’,7’-Dichlorofluorescin diacetate (DCF-DA) was obtained from Molecular Probes (Eugene, OR, USA). H2O2 was purchased from Acros Organics (Pittsburgh, PA, USA). The ECL detection system was from Amersham Pharmacia Biotech (Uppsala, Sweden). Bosentan (N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-pyrimidin-2-yl-pyrimidin-4-yl]-4-tert-butyl-benzenesulfonamide), a nonselective ETA/ETB receptor antagonist, was obtained from Actelion Pharmaceuticals (Allschwil, Switzerland). U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene) was obtained from Tocris Cookson Ltd (Bristol, UK). The plasmid AP-1-Luc containing the firefly luciferase reporter gene driven by a basic promoter element (TATA box) joined to tandem repeats of AP-1 binding element were obtained from Stratagene (La Jolla, CA, USA). Uric acid, N-acetylcysteine (NAC), apocynin, probenecid (4-[dipropylsulfamoyl]benzoic acid), benzbromarone ([3,5-dibromo-4-hydroxyphenyl]-[2-ethyl-3-benzofuranyl]methanone) and all other reagent-grade chemicals were purchased from the Sigma-Aldrich Chemical Co (St Louis, MO, USA).
Culture of human aortic smooth muscle cells and treatments Primary cultures of HASMC were obtained from PromoCell GmbH (Heidelberg, Germany) and cultured as per the manufacturer instructions. Cells were passaged every 3–4 d at a split ratio of 1:4 in growth medium (SMCGM2; Promocell). SMCGM2 is a commercially produced medium supplemented with 5% fetal calf serum (FCS), epidermal growth factor (EGF) (0.5 ng/mL), basic fibroblast growth factor (bFGF) (2 ng/mL), insulin (5 µg/mL), amphotericin B (50 ng/mL) and gentamicin (50 µg/mL). Cells were then treated with basal medium containing no serum or growth factors in the following experiments. All experiments were carried out between passages 5 and 16. For analysis of the effects of uric acid, cells were seeded in growth medium at a pre-optimized density of 5×103 cells/cm2 in 96-well, 12-well or 6-well plates or 60-mm culture dishes, as appropriate. After 3 d (about 70%–80% confluency) existing medium was replaced with basal medium for 24 h. Monolayers were then treated in fresh basal medium, alone (control) or with reagents as indicated. Inhibitor concentrations used were bosentan 3 or 30 µmol/L, U0126 10 µmol/L, probenecid 1 mmol/L and benzbromarone 50 µmol/L (30 min preincubation). Antioxidant concentrations were NAC 10 mmol/L and apocynin 10 µmol/L (30 min preincubation).
Cell proliferation Proliferation was assessed by counting and [3H]thymidine incorporation of cells incubated with or without uric acid, in the presence or absence of reagents as indicated. The rate of cellular proliferation was determined by cell counting. Cells were removed from the culture dish by addition of trypsin, and pelleted by centrifugation. The pellet was resuspended in 1 mL DMEM and cells were counted in an automatic cell counter (S.ST.II/ZM, Coulter Electronics Ltd, Miami, FL, USA). To measure synthesis of new DNA, cells were plated on 6-well (35 mm) dishes 24 h before experiments as previously described[12]. Cells were incubated with [3H]thymidine (5 µCi/mL). After addition of agent indicated, cells were harvested by incubation at 4 °C with trichloroacetic acid (5%) followed by solubilization in NaOH (0.1 mol/L), and radioactivity was determined by scintillation counting. Data are presented as the mean±SEM of 9–12 determinations in 3–4 different cell preparations and normalized to the untreated samplex100 (i.e. percentage of control).
Assay of ET-1 peptide secretion Endothelin-1 levels were measured in culture medium using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Amersham-Pharmacia, Amersham, UK) as previously described[12]. Results were normalized to cellular protein content in all experiments and expressed as a percentage relative to the cells incubated with the vehicle.
Detection of intracellular ROS and NADPH oxidase activity assay Reactive oxygen species were measured using a previously described method[12]. Prior to the chemical treatment, cells were incubated in culture medium containing a fluorescent dye, DCF-DA (30 µmol/L) for 1 h to establish a stable intracellular level of the probe. The same concentration of DCF-DA was maintained during the chemical treatment. Subsequently, the cells were washed with phosphate-buffered saline (PBS), removed from Petri dishes by brief trypsinization, and measured for 2’,7’-dichlorofluorescein (DCF) fluorescence intensity. The DCF fluorescence intensity of the cells is an index of intracellular levels of ROS, and it can be determined by fluorescence spectrophotometry with excitation and emission wavelengths at 475 and 525 nm, respectively. The cell number in each sample was counted in an automatic cell counter (S.ST.II/ZM, Coulter Electronics Ltd, Miami, FL, USA) and used to normalize the fluorescence intensity of DCF. Chemiluminescence assay of superoxide production was measured as described previously[14]. NADPH oxidase activity was measured as described previously[15]. NADPH oxidase activity was measured using the lucigenin-enhanced chemiluminescence method in microsomal membrane fractions. To prepare cell homogenates, the cell monolayer was washed three times with ice-cold PBS and scraped on ice in lysis buffer containing 20 mmol/L K-phosphate buffer (pH 7.0), 1 mmol/L glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), 1 mmol/L phenylmethanesulfonyl fluoride (PMSF), 10 µg/mL aprotinin, and 5 µg/mL leupeptin, followed by homogenization with 100 strokes in a Dounce homogenizer on ice. For the isolation of microsomal membranes, cell homogenates were prepared in 250 mmol/L sucrose, 5 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), 1 mmol/L PMSF, 10 µg/mL aprotinin, and 5 µg/mL leupeptin, followed by centrifugation at 1000×g (10 min, 4 °C). The pellet was discarded, and the supernatant was spun at 8000×g (10 min, 4 °C). The microsomal fraction was separated from cytosol by centrifugation of the supernatant at 105 000×g (45 min, 4 °C). The pellet was resuspended in the homogenization buffer by using a Hamilton glass syringe. The cell homogenate and microsomal fraction were used immediately. The assay was started in an Orion microplate luminometer (Berthold Detection Systems, GmbH of Pforzheim, Germany) by automatic injection of the 150 µL reaction buffer (50 mmol/L K-phosphate buffer [pH 7.0] containing 1 mmol/L EGTA, 150 mmol/L sucrose, 5 µmol/L lucigenin, and 100 µmol/L NADPH) into 10 µL of the homogenate or membrane suspension (5–20 µg protein). Photon emission in response to superoxide generation was measured every 60 s with a 5 s signal integration time for 20 min. The activity is expressed in relative light units per milligram of protein. The protein concentration was measured using the bicinchoninic acid protein assay (Pierce, Rockford, IL, USA).
RNA isolation and Northern blot analysis Total RNA was isolated from cells by the guanidine isothiocyanate/phenol chloroform method as previously described[12]. Blots were stripped and reprobed for 18S cDNA probe (obtained from American Type Culture Collection [Rockville, MD, USA]) to control for loading. Expression of ET-1 mRNA was quantitated and was normalized to the 18S signal.
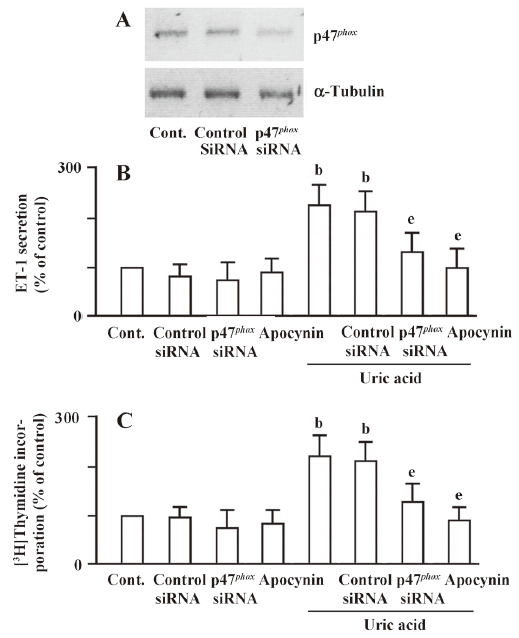
siRNA-mediated gene knockdown p47phox siRNA (sc-36157), and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and used for p47phox knockdown, and mock control, respectively[16]. Transfection of siRNA in HASMC was carried out using nucleofection according to the manufacturer’s instruction (Amaxa, Gaithersburg, MD, USA). Transfected cells were applied to advanced assays and Western blot analysis.
Western blot analysis Rabbit polyclonal anti-phospho-specific ERK antibodies were purchased from New England Biolabs (Beverly, MA, USA). Anti-ERK antibodies, anti-p47phox antibodies and anti-α tubulin antibodies were purchased from Santa Cruz Biotechnology. Western blot analysis was carried out as previously described[12]. Cells were rinsed twice with ice-cold PBS following treatment and scraped into ice-cold buffer containing 50 mmol/L Tris·HCl (pH 7.6), 120 mmol/L NaCl, 1% Nonidet P-40, 10% glycerol, 1 mmol/L PMSF, 2 mmol/L sodium orthovanadate, 10 mmol/L sodium pyrophosphate, 40 µg/mL leupeptin, 5 µg/mL aprotinin, 1 µg/mL pepstatin, 100 mmol/L NaF, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), and 1 mmol/L EGTA. After lysis on ice for 60 min, extracts were centrifuged for 10 min at 14 000×g at 4 °C. The protein concentration of the supernatants was measured, and 20 µg protein samples of cell lysate were mixed (1:1) with Laemmli sample buffer and incubated at 95 °C for 5 min. Proteins were resolved by spdium dodecyl sulfate-plyacrylamide gel electrophoresis (SDS-PAGE), followed by electroblotting onto polyvinylidene difluoride (PVDF) membrane. Membranes were blocked in 10 mmol/L Tris (pH 7.5), 100 mmol/L NaCl, and 0.1% Tween 20 containing 5% non-fat dry milk, followed by incubation with primary antibody. Membranes were washed 3 times and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody. The immunocomplexes were visualized by chemiluminescence with the Phototope Western blot detection system (Cell Signaling Technology, Beverly, Massachusetts, USA). The images were digitalized using the AlphaEase FluorChem digital imaging system (Alpha Innotech, San Leandro, CA, USA). Band densitometry was carried out using NIH Image software. Ratios of phosphorylated kinases to total kinases or to α tubulin were calculated.
Transfection and chloramphenicol acetyltransferase assays For the transient transfections, cells transfected with different expression vectors were obtained with the Amaxa Nucleofector system (Amaxa, Gaithersburg, MD, USA), which allows non-viral gene to be directly transferred into the nucleus using proprietary Nucleofector solutions. To correct for variability in transfection efficiency, 5 μg of pSV-β-galactosidase plasmid DNA was cotransfected in all of the experiments. The CAT and β-galactosidase assays were carried out as described previously[17]. The relative CAT activity was corrected by normalizing the respective CAT value to that of β-galactosidase activity. Cotransfected β-galactosidase activity varied by <10% within a given experiment and was not affected by any of the experimental manipulations described. As for positive and negative controls, pBLCAT2 (with thymidine kinase promoter) and pBLCAT3 (without promoter) were included in each assay.
Luciferase assay Human aortic smooth muscle cells plated on 6 well (35 mm) dishes were transfected with the luciferase reporter construct possessing consensus AP-1 binding sites (AP-1-Luc) (Stratagene). HASMC were assayed for luciferase activity with a luciferase reporter assay kit (Stratagene) as previously described[12]. The firefly luciferase activities as AP-1 transcriptional activity were normalized for transfection efficiency to its respective β-galactosidase activity and expressed as relative activity to control.
Statistical analysis Results are expressed as mean± SEM. Statistical analysis was carried out using Student’s t test and ANOVA followed by a Dunnett multiple comparison test using Prism version 3.00 for Windows (GraphPad Software, San Diego, CA, USA). A value of P<0.05 was considered to be statistically significant.
Results
Effect of uric acid on cell proliferation and ET-1 expression in human aortic smooth muscle cells The effect of uric acid on cell proliferation was evaluated by means of cell count and [3H]thymidine incorporation studies. Uric acid (300 and 600 µmol/L, 24 h) significantly increased cell number and DNA synthesis in HASMC (Figure 1A, 1B). Uric acid (300 µmol/L) also significantly increased the proliferative activity of HASMC in a time-dependent manner (Figure 1C, 1D). Uric acid (300 µmol/L, 24 h)-increased cell number and DNA synthesis in HASMC was inhibited by pan-ET-receptor antagonist bosentan (30 µmol/L) treatment (Figure 1E, 1F). These data suggest the possible role of endogenous ET-1 as an autocrine growth factor for the proliferation of HASMC under uric acid stimulation.
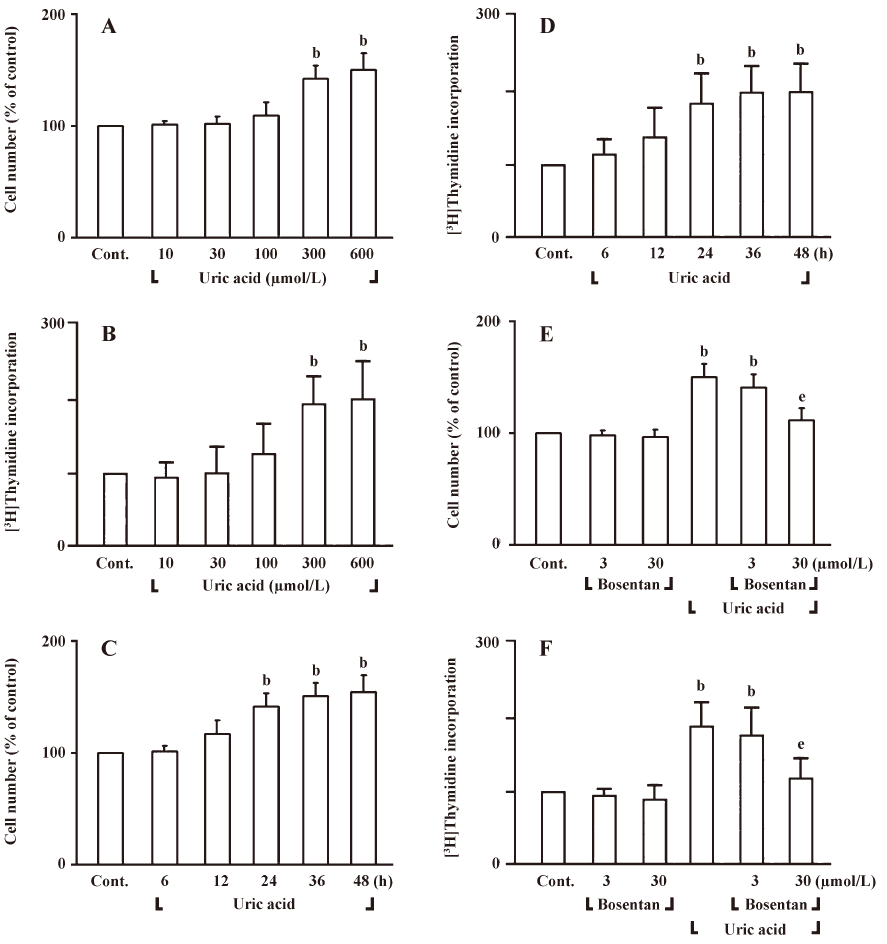
To determine whether uric acid would increase ET-1 mRNA levels in HASMC or not, we carried out Northern blot analysis (Figure 2A, B). In HASMC, uric acid induced ET-1 mRNA expression in a time-dependent manner (uric acid 300 µmol/L) (Figure 2A). When cells were treated with uric acid for 6 h, uric acid (100–600 µmol/L) significantly increased ET-1 mRNA expression (Figure 2B). To examine whether uric acid would increase ET-1 expression in HASMC, concentration-dependency of ET-1 secretion by uric acid had been carried out by ELISA (Figure 2C). HASMC exposed to 24 h of uric acid (300 and 600 µmol/L) significantly increased ET-1 peptide secretion (Figure 2C). To determine whether transport of uric acid into HASMC required the stimulation of ET-1 peptide secretion, we pretreated cells with probenecid and benzbromarone, two structurally unrelated organic anion transporter inhibitors of the transmembrane transport of urate[18]. As shown in Figure 2D, both inhibitors prevented stimulation of ET-1 peptide secretion in HASMC in response to uric acid, suggesting that uric acid must have entered the cell to induce ET-1 peptide secretion. These data show that uric acid induces cell proliferation and ET-1 gene expression in HASMC.
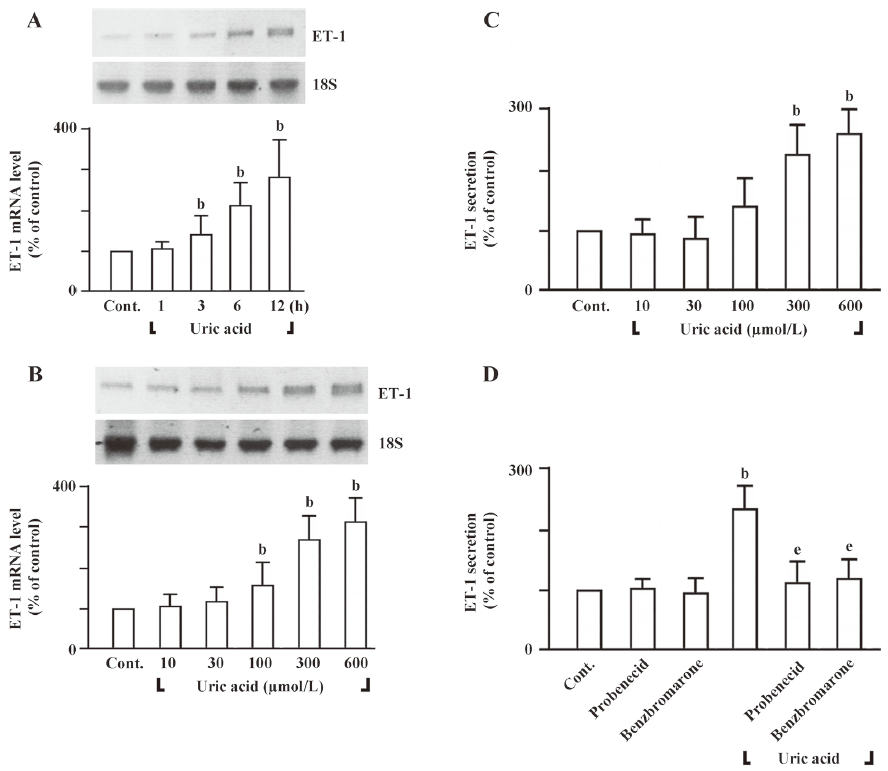
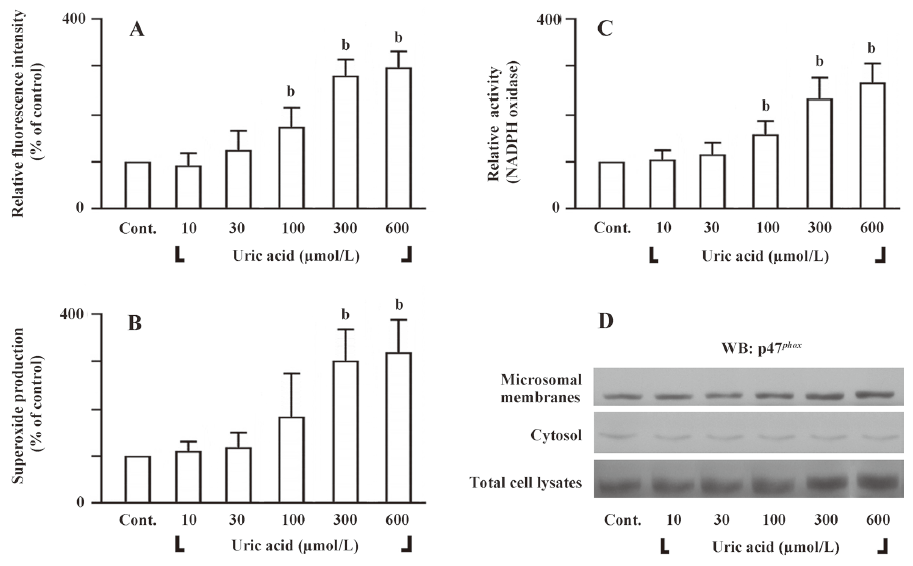
Uric acid-induced cell proliferation and ET-1 gene expression is redox-sensitive To determine whether uric acid would induce intracellular ROS in HASMC or not, we measured intracellular ROS levels by analyzing the fluorescent product DCF, a peroxidative product of DCF-DA. HASMC treated with uric acid (100–600 µmol/L) for 30 min had ROS levels significantly higher than those cells treated with vehicle only (Figure 3A). Similar results were obtained with the use of lucigenin-enhanced chemiluminescent superoxide detection (Figure 3B). Next, we examined the effect of uric acid on the enzymatic activity of NADPH oxidase. Since active NADPH oxidase is a membrane-associated enzyme[19], we tested the effect of uric acid on the NADPH oxidase activity in microsomal membranes. Treatment of HASMC with uric acid (100–600 µmol/L) for 30 min stimulated NADPH oxidase activity in the microsomal fraction (Figure 3C). NADPH oxidase is composed of several regulatory subunits, including those involved in the formation of the classic phagocyte-type NADPH oxidase (gp91phox, p67phox, p47phox, p40phox, and p22phox)[19]. We assessed the effect of uric acid on translocation of p47phox to the membranes. Immunoblot analysis of subcellular fractions of HASMC treated with uric acid revealed that uric acid (100–600 µmol/L; for 30 min) induced a dramatic dose-dependent increase in the content of p47phox in the microsomal fraction with a simultaneous decrease in the cytosol (Figure 3D), indicating translocation of the subunit from the cytosol to membranes.
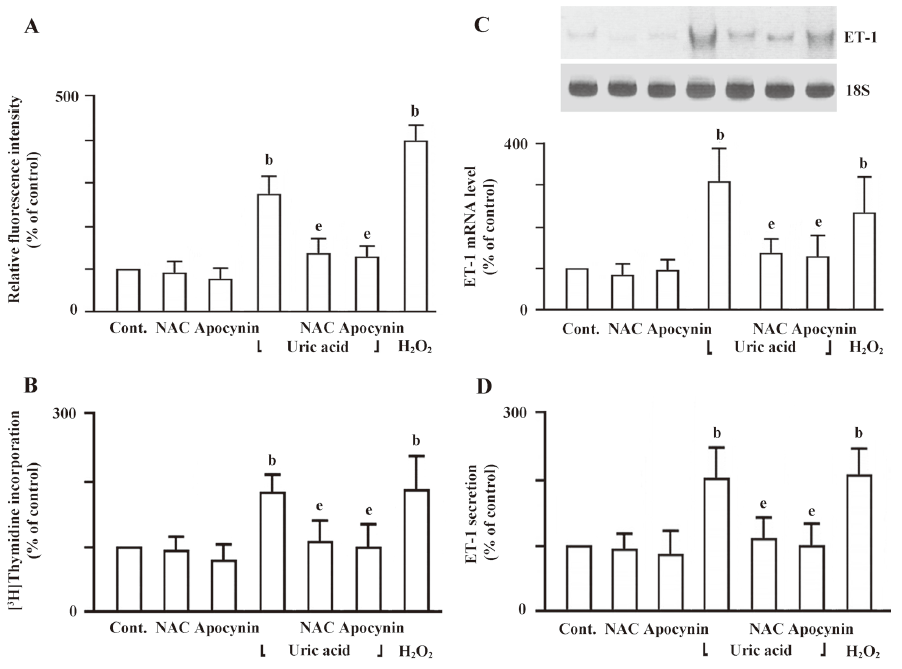
In addition, cells pretreated with antioxidants such as NAC (10 mmol/L) or a catechol-methyl derivative that blocks p47phox interaction with gp91phox/p22phox, apocynin (10 µmol/L) for 30 min showed a significant reduction in ROS production (Figure 4A). To elucidate the involvement of ROS in the uric acid-induced cell proliferation and ET-1 expression, HASMC were pretreated with NAC or apocynin for 30 min followed by uric acid treatment. HASMC pretreated with NAC or apocynin significantly suppressed uric acid-increased cell proliferation, ET-1 mRNA level, and ET-1 peptide secretion (Figure 4B, 4C, 4D). These findings suggest that intracellular ROS generation plays a role in uric acid-induced cell proliferation and ET-1 gene expression in HASMC.
Uric acid-induced cell proliferation and ET-1 gene expression is associated with NADPH oxidase To confirm the role of NADPH oxidase in uric acid-induced ET-1 expression and cell proliferation, NADPH oxidase subunit p47phox siRNAs were used for NADPH oxidase knockdown cells. As shown in Figure 5A, the expression level of p47phox was significantly reduced in p47phox siRNA transfection cells compared with the mock controls. The uric acid-induced ET-1 peptide secretion and cell proliferation were also decreased apparently in siRNA transfection cells (Figure 5B, 5C). These results revealed that uric acid induces ET-1 expression and cell proliferation via NADPH oxidase in HASMC.
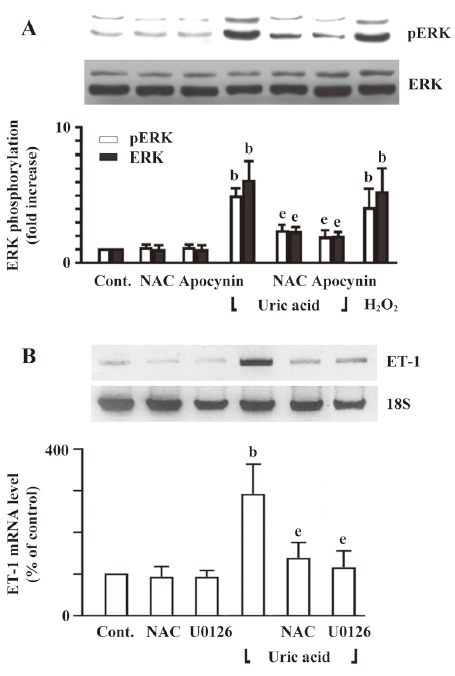
Involvement of ERK phosphorylation in uric acid-induced ET-1 gene expression To study the intracellular pathways that were involved in uric acid-induced ET-1 gene expression in HASMC, we examined the effect of uric acid on ERK phosphorylation and determined the effect of ERK inhibitors on uric acid-induced ET-1 gene expression. We found that uric acid increased ERK phosphorylation in HASMC (Figure 6A). Both NAC and apocynin significantly inhibited uric acid-induced ERK phosphorylation (Figure 6A). We next determined the role of redox-sensitive activation of ERK in uric acid-induced ET-1 gene expression. NAC or U0126 (10 µmol/L), an inhibitor of MKK-1 (MEK), inhibited the augmentation of ET-1 mRNA expression stimulated by uric acid (Figure 6B). These findings implicate the redox-sensitive activation of ERK signaling pathways in uric acid-induced ET-1 mRNA expression.
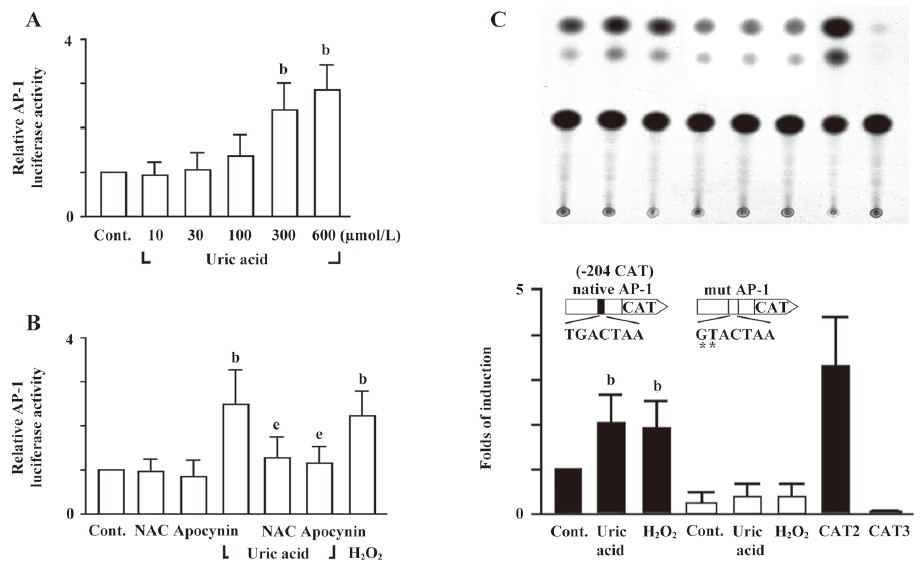
Identification of uric acid-responsive regulatory elements in the ET-1 promoter The ET-1 promoter contains an AP-1 site, which can be regulated by multiple activation pathways[20,21]. As shown in Figure 7A, uric acid (300 and 600 µmol/L) significantly increased AP-1-mediated reporter activity in HASMC. Moreover, pretreated cells with antioxidants, NAC or apocynin attenuated the uric acid-stimulated AP-1-mediated reporter activity (Figure 7B). We further examined whether the AP-1 site is essential for the induction of ET-1 gene by uric acid. In cells transfected with reporter construct -204CAT with two-bp mutation in the AP-1 site, the uric acid-induced ET-1 promoter activity was completely abolished. In addition, the basal promoter activity also decreased as compared with the control (Figure 7C). These findings suggest that the AP-1 binding element is essential for the induction of the ET-1 gene by uric acid. These results clearly indicate that ROS mediate the transcriptional activity of AP-1 induced by uric acid, and the AP-1 binding element is responsible for the induction of ET-1 gene expression by uric acid in HASMC.
Discussion
The results of the present study demonstrate that uric acid exerts direct effects on HASMC. In this study, we report that soluble uric acid stimulates cell proliferation and increases ET-1 expression in HASMC. The increase in ET-1 expression with both mRNA and protein were upregulated within a few hours of incubation of the HASMC with uric acid. The increase led us to examine the possible posttranscriptional effects of uric acid, as well as various signaling pathways and transcription factors known to be important in ET-1 regulation.
We observed concentration-dependent effects of uric acid in HASMC within a concentration range of 100–600 µmol/L, with distinct effects observed at concentrations as low as 300 µmol/L. It is important to note that uric acid levels in most mammals differ from that seen in humans. In humans, hyperuricemia is defined as uric acid levels >415 µmol/L in men and >360 µmol/L in women (with a normal range of ~120–415 µmol/L)[2], and has been identified as a risk factor in the development of hypertension[22,23].
Direct effects of soluble uric acid were characterized in various cell types, including vascular smooth muscle cells, endothelial cells, and adipocytes. The observation that ET-1 peptide secretion in response to uric acid is sensitive to blockade of urate transport with probenecid and benzbromarone suggests that entry of uric acid into the cell is required for these effects. In vascular smooth muscle cells, uric acid activates critical proinflammatory pathways[5] and stimulates cell proliferation[7]. In endothelial cells, uric acid decreases nitric oxide bioavailability and inhibits cell migration and proliferation, which are mediated in part by the expression of C-reactive protein[7]. In adipocytes, the redox-dependent effects of uric acid are mediated by the activation of intracellular oxidant production via NADPH oxidase[8]. Activation of ERK in response to uric acid has been shown in vascular smooth muscle cells and adipocytes[5,8]. In assessing the mechanism through which uric acid mediated its effects in HASMC, we also found that redox pathways were implicated. The vascular NAD(P)H oxidases have been found to be essential in the physiological and pathological responses of vascular cells, including growth, migration, and modification of the extracellular matrix[24–26].
Uric acid induced activation of NADPH oxidase in crude homogenates and isolated microsomal membranes of HASMC. Mechanisms of activation of NADPH oxidase (NOX) enzymes vary greatly depending on the spectrum of expressed isoforms and cytoplasmic regulators. NOX1–NOX3 require the presence of p22phox on the membrane and are more or less dependent on cytoplasmic subunits p47phox, p67phox, and p40phox, which translocate to the membrane and form with NOX and p22phox an active holoenzyme complex in response to cell stimulation[19]. These NOX isoforms can provide constitutive and stimulated ROS production in HASMC, and different mechanisms may be involved in the activation of NADPH oxidase in response to stimulation. Our data on translocation of p47phox in response to uric acid indicate the possibility of assembly of the classic phagocyte-type NADPH oxidase based on gp91phox or another NOX protein. The upstream signaling mechanism linking urate transport into the cell and formation of the active NOX complex in HASMC remains largely unknown. The p47phox plays a major role in functionally active NAD(P)H oxidase activity in cardiovascular cells, as evidenced by studies using p47phox–/– mice[27,28]. Furthermore, in vascular smooth muscle cells from human resistance arteries that c-Src-mediated phosphorylation of p47phox is a prerequisite for assembly and translocation of the p40-p47-p67phox complex[29]. The ability of NAC, and apocynin, which blocks association of p47phox with membrane-associated subunits[29,30], and NADPH oxidase subunit p47phox knockdown to inhibit the uric acid-induced increase in cell proliferation and ET-1 expression suggested that uric acid was acting in a pro-oxidative manner associated with NADPH oxidase. Uric acid is often thought of as a chemical antioxidant, and is considered to be a major antioxidant in the human plasma[1]; however, available data suggest that uric acid is not necessarily an antioxidant and, depending on the chemical milieu, may become a pro-oxidant[8]. Several studies consistent with our present findings have demonstrated that uric acid can be pro-oxidative and may even generate free radicals[5,8]. The ability of uric acid to activate ERK, and AP-1, as well as to increase ET-1 expression, is consistent with an oxidant-driven pathway.
Our results support previous epidemiological studies of hyperuricemia, which suggest an involvement of uric acid in the pathogenesis of the cardiovascular diseases, and provide a possible molecular mechanism for this role based on the finding that soluble uric acid affects HASMC directly by inducing ROS production. We suggest that hyperuricemia can be one of the causal factors inducing intracellular oxidative stress followed by a mitogenic process in the vascular tissue, thereby contributing to the pathogenesis of the cardiovascular diseases. However, further experiments will be necessary to identify the precise mechanisms by which uric acid is associated with vascular diseases. Thus, this study delivers important new insight into the molecular pathways that may contribute to the deleterious effects of uric acid in regard to hypertension and cardiovascular diseases.
Acknowledgements
This study was supported by grants from the National Science Council (NSC 93-2320-B-038-020-; NSC 94-2314-B-038-062-) and Shin Kong Wu Ho-Su Memorial Hospital (SKH-TMU-94-16), Taipei, Taiwan, China. The authors wish to thank Miss Sherry LO for assistance in manuscript editing and revision.
References
- Feig DI, Kang DH, Nakagawa T, Mazzali M, Johnson RJ. Uric acid and hypertension. Curr Hypertens Rep 2006;8:111-5.
- Gertzberg N, Neumann P, Rizzo V, Johnson A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol 2004;286:L37-48.
- Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 2006;8:691-728.
- Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001;38:1101-6.
- Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002;40:355-60.
- Itoh H, Higuchi H, Hiraoka N, Ito M, Konishi T, Nakano T, et al. Contraction of rat thoracic aorta strips by endothelin-1 in the absence of extracellular Ca2+. Br J Pharmacol 1991;104:847-52.
- Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol 2001;1:181-93.
- Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 2007;293:C584-96.
- Iglarz M, Schiffrin EL. Role of endothelin-1 in hypertension. Curr Hypertens Rep 2003;5:144-8.
- Ergul A. Endothelin-1 and endothelin receptor antagonists as potential cardiovascular therapeutic agents. Pharmacotherapy 2002;22:54-65.
- Suzuki S, Wenyi Z, Hirai M, Hinokio Y, Suzuki C, Yamada T, et al. Genetic variations at urotensin II and urotensin II receptor genes and risk of type 2 diabetes mellitus in Japanese. Peptides 2004;25:1803-8.
- Hong HJ, Chan P, Liu JC, Juan SH, Huang MT, Lin JG, et al. Angiotensin II induces endothelin-1 gene expression via extracellular signal-regulated kinase pathway in rat aortic smooth muscle cells. Cardiovasc Res 2004;61:159-68.
- Cheng TH, Shih NL, Chen SY, Loh SH, Cheng PY, Tsai CS, et al. Reactive oxygen species mediate cyclic strain-induced endothelin-1 gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in endothelial cells. J Mol Cell Cardiol 2001;33:1805-14.
- Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ Res 1997;81:1-7.
- Cheng JJ, Chao YJ, Wang DL. Cyclic strain activates redox-sensitive proline-rich tyrosine kinase 2 (PYK2) in endothelial cells. J Biol Chem 2002;277:48152-7.
- Chen CH, Cheng TH, Lin H, Shih NL, Chen YL, Chen YS, et al. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of Src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol Pharmacol 2006;69:1347-55.
- Cheng TH, Shih NL, Chen SY, Wang DL, Chen JJ. Reactive oxygen species modulate endothelin-I-induced c-fos gene expression in cardiomyocytes. Cardiovasc Res 1999;41:654-62.
- Miyazaki H, Sekine T, Endou H. The multispecific organic anion transporter family: properties and pharmacological significance. Trends Pharmacol Sci 2004;25:654-62.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
- Kawana M, Lee ME, Quertermous EE, Quertermous T. Cooperative interaction of GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol Cell Biol 1995;15:4225-31.
- Cheng TH, Cheng PY, Shih NL, Chen IB, Wang DL, Chen JJ. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol 2003;42:1845-54.
- Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension 2003;42:247-52.
- Cannon RO 3rd. Role of nitric oxide in cardiovascular disease: focus on the endothelium. Clin Chem 1998;44:1809-19.
- Grote K, Ortmann M, Salguero G, Doerries C, Landmesser U, Luchtefeld M, et al. Critical role for p47phox in renin-angiotensin system activation and blood pressure regulation. Cardiovasc Res 2006;71:596-605.
- Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 2003;24:471-8.
- Keaney JF Jr. Oxidative stress and the vascular wall: NADPH oxidases take center stage. Circulation 2005;112:2585-8.
- Lavigne MC, Malech HL, Holland SM, Leto TL. Genetic demonstration of p47phox-dependent superoxide anion production in murine vascular smooth muscle cells. Circulation 2001;104:79-84.
- Cheng CM, Hong HJ, Liu JC, Shih NL, Juan SH, Loh SH, et al. Crucial role of extracellular signal-regulated kinase pathway in reactive oxygen species-mediated endothelin-1 gene expression induced by endothelin-1 in rat cardiac fibroblasts. Mol Pharmacol 2003;63:1002-11.
- Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2003;23:981-7.
- Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 2002;90:1205-13.