
HDT-1, a new synthetic compound, inhibits glutamate release in rat cerebral cortex nerve terminals (synaptosomes)1
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS) and plays an important role in many brain functions[1,2]. Apart from its normal physiological role, however, glutamate has also been suggested to be involved in the pathophysiology of a number of CNS disorders, especially when an imbalance in glutamatergic neurotransmission occurs like excessive glutamate release[3,4]. Therefore, inhibition of glutamate release is considered to be a potential mechanism for neuroprotective effects.
HDT-1 (3, 4, 4a, 5, 8, 8a-hexahydro-6,7-dimethyl-4a-(phenylsulfonyl)-2-tosylisoquinolin-1(2H)-one), a new synthetic compound[5], is similar in chemical structure to isoquinoline alkaloid, which has a remarkable spectrum of biological activities such as antioxidative effect[6]. Compounds that possess anti-oxidative activity might offer benefits in preventing or retarding some CNS disorders, as free radical-induced oxidative damage to the brain has been regarded as an important cause of neuronal death in a variety of neurodegenerative disorders of the CNS, as well as in ischemia and in the aging process[7,8]. Furthermore, this oxidative injury is reported to be associated with an excess release of glutamate[9,10].
Based on the above-mentioned fact, we hypothesized that HDT-1 could influence the release of glutamate. To address this hypothesis, therefore, the present study was designed to use isolated nerve terminals (synaptosomes) prepared from the rat cerebral cortex to investigate the effect of HDT-1 on glutamate release. The synaptosome preparation provides a useful system for studying the specific presynaptic regulation of neurotransmitter release by a drug, given that the preparation is devoid of functional glial and nerve cell body elements that might obfuscate interpretation because of modulatory loci at non-neuronal, postsynaptic, or network levels[11]. Measurement of glutamate release with synaptosomes is a good model to quickly screen new compounds as well as to investigate the mechanisms underlying the action of compounds. Here, we find that HDT-1 potently inhibits depolarization-evoked glutamate release from cerebrocortival nerve terminals. This effect is associated with a reduction of voltage-dependent Ca2+ channel activity and the subsequent decrease of Ca2+ influx into nerve terminals, rather than any upstream effect on nerve terminal excitability.
Materials and methods
Materials HDT-1 was synthesized by Prof Shang-shing P CHOU (Department of Chemistry) and was generously given to us. The structure of HDT-1 is shown in Figure 1. 3’,3’,3’-dipropylthiadicarbocyanine iodide (DiSC3(5)) and Fura-2-acetoxymethyl ester (Fura-2-AM) were obtained from Molecular Probes (Eugene, OR, USA). ω-Conotoxin MVIIC (ω-CgTX MVIIC) and dantrolene were obtained from Tocris Cookson (Bristol, USA). BAPTA-AM, and all other reagents were obtained from Sigma (Poole, UK) or Merck (Poole, UK).
Isolation of synaptosomes from rat cerebral cortex All experiments were carried out in accordance with the guidelines established by the Fu Jen Institutional Animal Care and Utilization Committee. Synaptosomes were purified by discontinuous Percoll gradients as described previously[12]. The cerebral cortex from male Sprague-Dawley rats (2–3 months old) was isolated and homogenized in a medium containing 0.32 mol/L sucrose, pH 7.4. The homogenate was centrifuged at 3000×g for 2 min at 4 °C. The supernatant fraction was collected and centrifuged at 14 500×g for 12 min. The resulting pellet was resuspended in 8 mL of 0.32 mol/L sucrose, pH 7.4. 2 µL of this synaptosomal suspension were loaded onto discontinuous gradients consisting of three 2-mL layers of filtered Percoll density gradient medium (23%, 10%, and 3%) in 0.32 mol/L sucrose containing 0.25 mmol/L dithiothreitol and 1 mmol/L ethylenediaminetetraacetic acid, pH 7.4. The gradients were centrifuged at 32 500×g for 7 min at 4 °C. The synaptosomal fraction was collected from the 23%/10% Percoll interface and diluted in a volume of 30 mL of HEPES buffer medium consisting of 140 mmol/L NaCl, 5 mmol/L KCl, 5 mmol/L NaHCO3, 1.2 mmol/L NaH2PO4, 1 mmol/L MgCl2, 10 mmol/L glucose and 10 mmol/L HEPES (pH 7.4). The pellets were centrifuged at 27 000×g for 10 min and resuspended in the appropriate HEPES buffer medium to remove Percoll. The protein concentration of the synaptosomal preparation was determined by the method of Bradford using bovine serum albumin (BSA) as a standard. 0.5 mg of the synaptosomal suspension was diluted in 10 mL HEPES buffer medium and centrifuged at 3000×g for 10 min. The supernants were discarded and the pellets containing the synaptosomes were stored on ice and used within 4−6 h.
Measurement of glutamate release Glutamate release was assayed by on-line fluorimetry as described previously[12]. Synaptosomal pellets (0.5 mg/mL) were resuspended in 2 mL of HEPES buffer medium containing 16 µmol/L BSA and incubated in a stirred and thermostated cuvette maintained at 37 °C in a Perkin-Elmer LS-50B spectrofluorometer (Beaconsfield, UK). NADP+ (2 mmol/L), glutamate dehydrogenase (50 U/mL) and CaCl2 (1 mmol/L) were added after 3 min. After a further 5 min of incubation, 4-AP (3 mmol/L), or KCl (15 mmol/L) was added to stimulate glutamate release. The oxidative decarboxylation of released glutamate, leading to the reduction of NADP+, was monitored by measuring NADPH fluorescence at excitation and emission wavelengths of 340 nm and 460 nm, respectively. Data points were obtained at 2.2-s intervals. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment and the fluorescence change produced by the standard administration was used to calculate the released glutamate as nmol glutamate/mg synaptosomal protein. Release traces were shifted vertically to align the point of depolarization as zero release. Unless otherwise indicated, release values quoted in the text are levels attained at steady-state after 5 min of depolarization (nmol/mg per 5 min).
Synaptosomal membrane potential measurement The synaptosomal membrane potential can be monitored by positively charged membrane potential-sensitive carbocyanine dyes such as DiSC3(5)[13]. The dye becomes incorporated into the synaptosomal plasma membrane lipid bilayer. Upon depolarization with 4-AP, the release of the dye from the membrane bilayer is indicated as an increase in fluorescence. Synaptosomes were resuspended in 2 mL HEPES-buffered incubation medium and incubated in a stirred and thermostated cuvette at 37 °C in a Perkin-Elmer LS-50B spectrofluorometer (Beaconsfield, UK). After 3 min incubation, 4 µmol/L DiSC3(5) was added and allowed to equilibrate before the addition of CaCl2 (1 mmol/L) after 4 min incubation. Then, 4-AP was added to depolarize the synaptosomes at 10 min, and DiSC3(5) fluorescence was monitored at excitation and emission wavelengths of 646 nm and 674 nm, respectively, and data points were collected at 2.2-s intervals. Cumulative data were analysed using Lotus 1-2-3 and the results are expressed in fluorescence units.
Cytosolic Ca2+ measurement Synaptosomes (0.5 mg/mL) were preincubated in HEPES buffer medium with 16 µmol/L BSA in the presence of 5 µmol/L Fura-2-acetoxymethyl ester and 0.1 mmol/L CaCl2 for 30 min at 37 °C in a stirred test tube. After Fura-2-AM loading, synaptosomes were centrifuged in a microcentrifuge for 30 s at 3000×g. The synaptosomal pellets were resuspended in HEPES buffer medium with BSA and the synaptosomal suspension stirred in a thermostatted cuvette in a Perkin-Elmer LS-50B spectrofluorimeter. CaCl2 (1 mmol/L) was added after 3 min and further additions were made after an additional 5 min, as described in the legends of the figures. Fluorescence data were accumulated at excitation wavelengths of 340 nm and 380 nm (emission wavelength 505 nm) at data accumulated at 7.5-s intervals. Calibration procedures were carried out as described previously[14], using 0.1% SDS to obtain the maximal fluorescence with Fura-2 saturation with Ca2+, followed by 10 mmol/L egtazic acid (EGTA) (Tris buffered) to obtain minimum fluorescence in the absence of any Fura-2/Ca2+ complex. Cytosolic free Ca2+ concentration ([Ca2+]c, nmol/L) was calculated using equations described previously[15].
Statistical analysis Cumulative data were analysed using Lotus 1-2-3 and MicroCal Origin. Data are expressed as mean±SEM. To compare the difference of the effect of the drug treated group from that of the control group, a two-tailed Student’s t test was used. When an additional comparison was required (such as whether a second treatment influenced the actions of HDT-1), a one-way repeated-measures ANOVA was computed. P<0.05 was considered to represent a significant difference.
Results
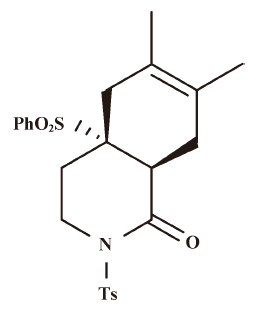
Effect of HDT-1 on glutamate release evoked by 4-AP and KCl To investigate the influence of HDT-1 on glutamate release, isolated nerve terminals were depolarized with the potassium channel blocker 4-aminopyridine (4-AP) or high external [K+]. 4-AP destabilizes the membrane potential and is thought to cause repetitive spontaneous Na+ channel-dependent depolarization that closely approximates in vivo depolarization of the synaptic terminal that leads to the activation of voltage-dependent Ca2+ channels (VDCC) and neurotransmitter release[16]. Elevated extracellular KCl concentrations depolarize the plasma membrane by shifting the K+ equilibrium potential above the threshold potential for activation of voltage-dependent ion channels. Although Na+ channels are inactivated under these conditions, VDCC are activated nonetheless to mediate Ca2+ entry supporting neurotransmitter release[17]. Under synaptosomes incubated in the presence of 1 mmol/L CaCl2, 4-AP (3 mmol/L) evoked a glutamate release of 7.8±0.1 nmol/mg per 5 min. Application of HDT-1 (10 µmol/L) produced an inhibition of 4-AP-evoked glutamate release to 4.5±0.3 nmol/mg per 5 min (n=8; P<0.01), without altering the basal release of glutamate (Figure 2A). Similarly, 15 mmol/L KCl evoked a control release of 9.3±0.1 nmol/mg per 5 min, which was also inhibited to 5.6±0.5 nmol/mg per 5 min in the presence of 10 µmol/L HDT-1 (n=8; P<0.01; Figure 2B). The HDT-1-induced inhibition of 4-AP- or KCl-evoked glutamate release was concentration-dependent, with an IC50 value derived from a dose-response curve of about 48 µmol/L and 61 µmol/L, respectively (Figure 2C). Given the robust depression of glutamate release seen with 10 µmol/L HDT-1, this concentration of HDT-1 was used in subsequent experiments to evaluate the mechanisms underlying the ability of HDT-1 to reduce glutamate release.
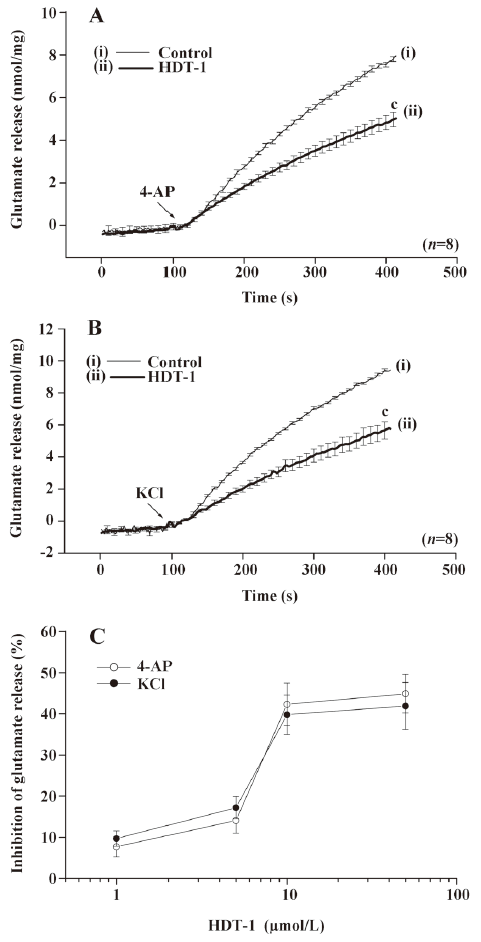
The data given above suggest that the inhibition by HDT-1 primarily results from a decrease in the activity of the release-coupled Ca2+ channels, because KCl-evoked glutamate release involves Ca2+ channels exclusively and 4-AP-evoked release involves both Na+ and Ca2+ channel activation. To further confirm this hypothesis, we examined the Ca2+-dependency of the 4-AP-evoked release. In the extracellular Ca2+-free medium containing 50 µmol/L BAPTA-AM, a cell-permeable Ca2+ chelator, the release of glutamate evoked by 4-AP (3 mmol/L) was 2.7±0.1 nmol/mg per 5 min. However, this Ca2+-independent glutamate release evoked by 4-AP was not significantly affected by HDT-1 (10 µmol/L) (2.5±0.3 nmol/mg per 5 min; n=6; Figure 3), suggesting that the inhibition of glutamate release by HDT-1 reflects the effects on the Ca2+-dependent exocytosis of glutamate.
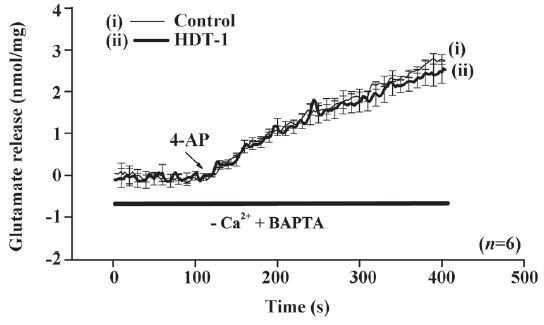
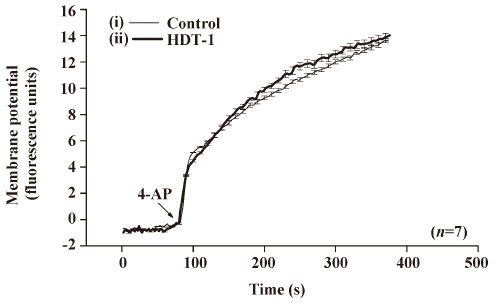
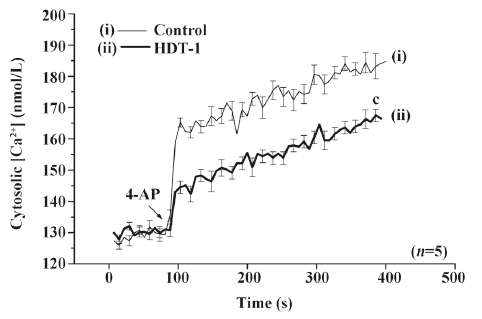
To further identify the mechanism behind the HDT-1-mediated inhibition of glutamate release, we assessed the effect of HDT-1 on synaptosomal plasma membrane potential and Ca2+ influx under resting conditions and on depolarization. Synaptosomal plasma membrane potential and depolarization was determined with the membrane potential-sensitive dye DiSC3(5). 4-AP (3 mmol/L) caused an increase in DiSC3(5) fluorescence of 13.6±0.2 fluorescence units/5 min. Preincubation of synaptosomes with 10 µmol/L HDT-1 for 10 min before 4-AP addition did not alter the resting membrane potential and produced no significant change in the 4-AP-mediated increase in DiSC3(5) fluorescence (13.9±0.3 fluorescence units/5 min; n=7; Figure 4). This suggests that the observed inhibition of 4-AP-evoked glutamate release by HDT-1 is unlikely to be due to a hyperpolarizing effect of the drug on the synaptosomal plasma membrane potential or an attenuation of depolarization produced by 4-AP. To investigate whether the inhibitory effect of HDT-1 on glutamate release reflected a decrease in Ca2+ influx, we used the Ca2+ indicator Fura-2 to assess the effect of HDT-1 on the 4-AP-induced increase in cytosolic Ca2+ levels ([Ca2+]c). As shown in Figure 5, after the addition of 4-AP (3 mmol/L), [Ca2+]c in synaptosomes was elevated to a plateau level of 184.8±3.2 nmol/L. Preincubation of synaptosomes with 10 µmol/L HDT-1 reduced the 4-AP-evoked [Ca2+]c increase by 12.4% (161.9±2.1 nmol/L; n=5; P<0.01).
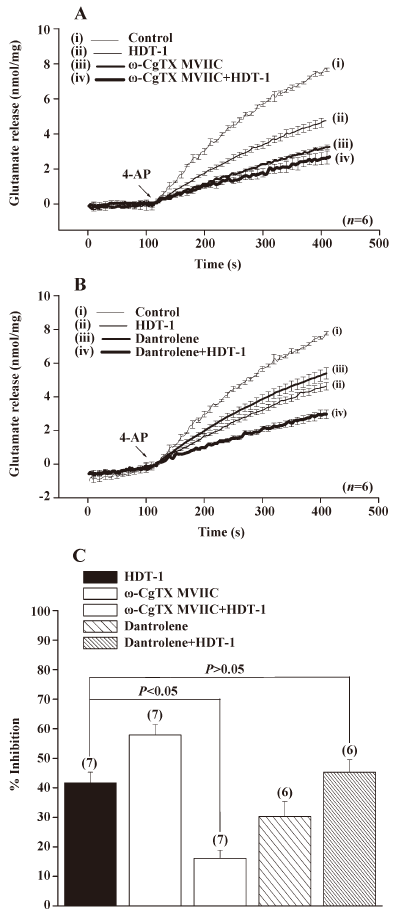
The data point to HDT-1 acted to affect voltage-dependent Ca2+ influx, rather than upstream loci in the stimulus-release cascade. Glutamate release from nerve terminals is supported by the entry of Ca2+ through the N- and P/Q-type Ca2+ channels[18,19]. To determine whether the modulation of these Ca2+ channel activities was involved in the HDT-1-mediated inhibition of glutamate release, we examined the effect of HDT-1 in the presence of ω-conotoxin MVIIC (ω-CgTX MVIIC), a wide spectrum blocker of N-, P-, and Q-type Ca2+ channels. As shown in Figure 6A, the application of 1 µmol/L ω-CgTX MVIIC caused a 59.8%±5.1% inhibition on 4-AP (3 mmol/L)-evoked glutamate release, from 7.7±0.1 nmol/mg per 5 min in control conditions to 3.1±0.1 nmol/mg per 5 min in the presence of ω-CgTX MVIIC. In the presence of ω-CgTX MVIIC, HDT-1 (10 µmol/L) inhibited 4-AP (3 mmol/L)-evoked glutamate release by only 16.1%±2.8% (2.6±0.3 nmol/mg per 5 min) compared with the control after the application of 1 µmol/L ω-CgTX MVIIC, which was significantly less than that when HDT-1 was applied alone (41.6%±3.7%; n=7; P<0.05; Figure 6C). In addition, we also tested the effect of dantrolene (100 µmol/L), an intracellular Ca2+ release inhibitor, on the action of HDT-1. In the presence of dantrolene (100 µmol/L), although control 4-AP (3 mmol/L)-evoked release was reduced to 5.3±0.3 nmol/mg per 5 min, HDT-1 (10 µmol/L) was still able to inhibit 4-AP (3 mmol/L)-evoked glutamate release by a further 45.3%±4.2% (2.9±0.2 nmol/mg per 5 min) (n=6; P<0.05; Figure 6B). Overall, these data point to the possibility that the effect of HDT-1 is manifested through the direct attenuation of presynaptic N- and P/Q-type Ca2+ channel activity.
Discussion
The release of neurotransmitter from the presynaptic site is a possible target for drug modulation of excitability and synaptic transmission in central neurons. By using a preparation of nerve terminals from rat cerebral cortex and by measuring the release of glutamate, we found that HDT-1, a synthetic compound, inhibits depolarization-evoked glutamate release. The mechanism of action involved in this release inhibition was investigated.
In principle, neurotransmitter release can be modulated at several loci in the stimulus-exocytosis cascade, including ion-channels modulating nerve terminal excitability, voltage-dependent Ca2+ channels, downstream of Ca2+ entry, components of the synaptic vesicle trafficking and exocytotic apparatus[20–22]. Thus, addressing the mechanism responsible for HDT-1-mediated inhibition of glutamate release, we considered 3 possibilities that might be involved: (1) an alteration of synaptosomal excitability,; (2) a direct inhibition of the exocytosis-coupled Ca2+ channels; and/or (3) a direct effect on some component of the release machinery. The first possibility appears unlikely on the basis of the following observations. First, HDT-1 significantly inhibited the release of glutamate evoked by 4-AP and KCl. Both of these depolarizing treatments are thought to activate N- and P/Q-type VDCC coupled to glutamate similarly and thus should reflect this by qualitatively similar modulation, if this occurs at the level of VDCC. Where the 2 depolarizing paradigms differ is in that, whereas 4-AP-mediated VDCC activation/glutamate release involves upstream ion (K+ and Na+)-channel activity, 15 mmol/L external KCl-induced VDCC activation/glutamate release involves only Ca2+ channels[17,23]. Based on the mechanistic differences between the 4-AP and the KCl-mediated depolarization discussed above, the data presented indicate that the suppression of glutamate release by HDT-1 reflects a direct effect on VDCC function. Second, no significant effect of HDT-1 on synaptosomal plasma membrane potential, measured with a membrane potential-sensitive dye DiSC3(5), was observed either under resting conditions or on depolarizing with 4-AP. Third, HDT-1 did not affect the 4-AP-evoked cytosolic efflux of glutamate in the absence of Ca2+, a component of glutamate release that is dependent only on membrane potential[12]. Therefore, these results suggest that the decreased glutamate release seen with HDT-1 is not due to a result of reducing the level of membrane depolarization.
On the other hand, our demonstration, using the Ca2+ indicator Fura-2, that the 4-AP-evoked increase in [Ca2+]c was reduced by HDT-1 suggests that observed HDT-1-mediated inhibition of glutamate release is indeed related to a decrease in the influx of Ca2+ in the cerebrocortical nerve terminals. In the rat cerebrocortical nerve terminal preparation, the release of glutamate depends on the Ca2+ influx through N- and P/Q-type Ca2+ VDCC[19]. Thus, we further examined whether Ca2+ entry specifically through N- and P/Q-type Ca2+ VDCC subserves the effect of HDT-1 by assessing the effect of HDT-1 in the presence of ω-CgTX MVIIC, a wide spectrum blocker of N-, P-, and Q-type Ca2+ channels. The finding that the inhibitory effect of HDT-1 on 4-AP-evoked glutamate release was significantly prevented by ω-CgTX MVIIC. Additionally, we found that HDT-1 continued to inhibit significantly 4-AP-evoked release of glutamate in the presence of dantrolene, an intracellular Ca2+ release inhibitor. Thus, these data point to the suppression of presynaptic N- and P/Q-type Ca2+ VDCC as the mechanism underlying the inhibition of glutamate release by HDT-1. However, the combined inhibition of N and P/Q-type Ca2+ channel activity could not fully block the action of HDT-1 on glutamate release (~6% of inhibition remained); thus, we cannot rule out the possibility that other unidentified types of Ca2+ channels or other presynaptic pathways might be involved in the action of HDT-1. Although the data presented suggest that HDT-1 limits glutamate release by reducing VDCC activity, a direct action at the exocytotic machinery itself downstream of Ca2+ entry could potentially inhibit release[24]. Whether HDT-1 could also modulate the release machinery remains unknown. Additional studies are required to test this possibility.
In conclusion, the results of this study demonstrate that, in cerebrocortical nerve terminals, HDT-1 inhibits evoked glutamate release by a reduction of Ca2+ influx through N- and P/Q-type Ca2+ channels, in the absence of any effect on nerve terminal excitability. As described in the introduction, excessive glutamate release resulting in neurotoxic cell damage has been implicated in the etiology of several neurological disease states, including ischemic brain damage and epilepsy[25,26]. Thus, the ability of HDT-1 to inhibit glutamate release suggests HDT-1 may serve as a potential neuroprotective agent. Although the mechanisms of the neuroprotection of HDT-1 are still under investigation, this finding may provide an important template for the development of novel therapeutically useful agents for the treatment of a wide range of neurological and neurodegenerative disorders.
Acknowledgements
We thank Jia-yu CHEN for expert technical assistance.
References
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem 1984;42:1-11.
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993;361:31-9.
- Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 1990;11:379-87.
- Obrenovitch TP, Urenjak J. Altered glutamatergic transmission in neurological disorders: from high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol 1997;51:39-87.
- Chou SSP, Chen PW. Cycloaddition reactions of 4-sulfur-substituted dihydro-2-pyridones and 2-pyridones with conjugated dienes. Tetrahedron 2008;64:1879-87.
- Cassels BK, Asencio M, Conget P, Speisky H, Videla LA, Lissi EA. Structure-antioxidative activity relationships in benzylisoquinoline alkaloids. Pharmacol Res 2004;31:103-7.
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993;262:689-95.
- Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Rad Biol Med 1997;23:134-47.
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci 1990;13:171-82.
- Tretter L, Adam-Vizi V. Glutamate release by an Na+ load and oxidative stress in nerve terminals: relevance to ischemia/reperfusion. J Neurochem 2002;83:855-62.
- Nicholls DG. The glutamatergic nerve terminal. Eur J Biochem 1993;212:613-31.
- Nicholls DG, Sihra TS, Sanchez-Prieto J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J Neurochem 1987;49:50-7.
- Akerman KE, Scott IG, Heikkila JE, Heinonen E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiSC2(5). J Neurochem 1987;48:552-9.
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985;260:3440-50.
- Sihra TS, Bogonez E, Nicholls DG. Localized Ca2+ entry preferentially affects protein dephosphorylation, phosphorylation, and glutamate release. J Biol Chem 1992;267:1983-9.
- Tibbs GR, Barrie AP, Van Mieghem FJ, McMahon HT, Nicholls DG. Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: effects on cytosolic free Ca2+ and glutamate release. J Neurochem 1989;53:1693-9.
- Barrie AP, Nicholls DG, Sanchez-Prieto J, Sihra TS. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J Neurochem 1991;57:1398-404.
- Turner TJ, Dunlap K. Pharmacological characterization of presynaptic calcium channels using subsecond biochemical measurements of synaptosomal neurosecretion. Neuropharmacology 1995;34:1469-78.
- Vazquez E, Sanchez-Prieto J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur J Neurosci 1997;9:2009-18.
- Roeper J, Pongs O. Presynaptic potassium channels. Curr Opin Neurobiol 1996;6:338-41.
- Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci 1993;16:222-7.
- Sihra TS, Nichols RA. Mechanisms in the regulation of neurotransmitter release from brain nerve terminals: current hypotheses. Neurochem Res 1993;18:47-58.
- Nicholls DG. Presynaptic modulation of glutamate release. Prog Brain Res 1998;116:15-22.
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 2004;27:509-47.
- Choi DW. Excitotoxic cell death. J Neurobiol 1992;23:1261-76.
- Lipton SA, Rosenberg PA. Excitatory amino acid as a final common pathway for neurologic disorders. N Engl J Med 1994;330:613-22.