
Role of rat liver cytochrome P450 3A and 2D in metabolism of imrecoxib1
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for the clinical treatment of pain, inflammation and fever[1,2]. These drugs (eg indomethacin, diclofenac and meloxicam) are thought to act by inhibiting cyclooxygenase (COX), an enzyme that limits the biosynthesis rate of prostaglandins from arachidonic acid[3–5]. Two forms of cyclooxygenase, designated COX-I and COX-II, exist[6,7]. COX-I is the major form, and is located in healthy tissues, catalyzes the formation of prostaglandins under normal physiological conditions, and plays a role in the maintenance of the gastrointestinal mucosa as well as platelet function[8]. In comparison, COX-II is an inducible enzyme that is predominantly expressed in association with inflammation[9]. It is believed that NSAID-induced gastrointestinal damage results from the inhibition of COX-I, whereas the therapeutic benefit results from the inhibition of COX-II expressed at the site of inflammation[10]. However, the division between the biological functions of COX-I and COX-II is not clear-cut. Moreover, treatment with COX-II selective inhibitors could theoretically lead to problems with thrombosis, and salt and water balance. Recently, much attention has been focused on the increased risk of cardiovascular events associated with COX-II selective NSAIDs (such as celecoxib and rofecoxib) as compared with nonselective NSAIDs[11–13]. Therefore, NSAIDs that preferentially inhibit COX-II with moderate selectivity seem more promising. Imrecoxib, [4-(4-methane-sulfonyl-phenyl)-1-propyl-3-p-tolyl-1,5-dihydro-pyrrol-2 -one] (Figure 1), is a novel and moderately selective COX-II inhibitor[14–16]. The drug inhibits COX-I and COX-II with IC50 values of 115±28 nmol/L and 18±4 nmol/L, respec-tively. Imrecoxib exerts its anti-inflammatory effect by inhibiting COX-II mRNA expression[16]. In fact, imrecoxib is currently undergoing clinical trials in China for the treatment of acute and chronic inflammatory disease. We previously found that imrecoxib is extensively metabolized in rats, with less than 2% of the dose excreted unchanged in urine and feces (unpublished data). We characterized 7 metabolites of imrecoxib in rats: the 4'-hydroxymethyl (M4), 4'-carboxylic acid (M2), 4'-hydroxymethyl-5-hydroxy (M3), and 4'-hydroxymethyl-5-carbonyl (M5) metabolites, and glucuronide conjugates of M2, M3, and M4. The major route of metabolism appears to be 4'-methyl hydroxylation, with further oxidation of the corresponding carboxylic acid (Figure 1). The purpose of the present study was to study the in vitro metabolism of imrecoxib in rat liver microsomes and to identify the cytochrome P450 (CYP) forms involved in its metabolism.

Materials and methods
Chemicals Imrecoxib, the 4'-hydroxymethyl (M4) and 4'-carboxylic acid (M2) metabolites of imrecoxib, and BAP 910 (an analogue of imrecoxib, used as internal standard) were supplied by the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). 4'-Hydroxymethyl-5-hydroxyl (M3) and 4'-hydroxy-methyl-5-carbonyl (M5) metabolites of imrecoxib, used as reference substances, were isolated from rat urine in our laboratory and identified by mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy. α-Naphthoflavone, β-naphthoflavone, methylpyrazole, quinine, codeine, and morphine were all purchased from Sigma-Aldrich (Deisenhofen, Germany). Cimetidine was obtained from Kaili Pharmaceutical Co (Jiangsu, China), ketoconazole from Dragon Pharmachemical Co (Zhejiang, China), dexamethasone from Tianjin Pharmaceutical Group Co (Tianjin, China), isoniazid from Jiangbei Pharmaceutical Co (Zhejiang, China), nifedipine from Zhongnuo Pharmaceutical Co (Shijiazhuang, China), diphenhydramine from Beijing Taiyang Pharmaceutical Co (Beijing, China), and gliclazide from Tianjin Zhongxin Pharmaceutical Co (Tianjin, China). Dehydronifedipine was synthesized in the Department of Pharmaceutical Chemistry, Shenyang Pharmaceutical University (Shenyang, China). β-Nicotinamine adenine dinucleotide phosphate (reduced form, NADPH) was obtained from Xinjingke Biotechnology Co (Beijing, China). DL-Dithio-threitol (DTT, ultrapure grade) and tris (hydroxymethyl)aminomethane (Tris, ultrapure grade) were obtained from Ameresco (Solon, Ohio, USA). Methanol and acetonitrile were of high performance liquid chromatography grade (Yuwang Co, Shandong, China). All other chemicals were of analytical grade.
Animal preparation Male Wistar rats (7*8 weeks old) weighing 200 to 250 g were supplied by the Laboratory Animal Center of Shenyang Pharmaceutical University (grade II, certificate N
Preparation of rat liver microsomes Pooled liver microsomes from 6 rat livers in each group were prepared as previously described[17]. The microsomal protein concentrations were determined by using the method of Lowry et al[18]. Total cytochrome P450 contents were measured according to the method of Omura and Sato[19].
Incubation of imrecoxib with rat liver microsomes To determine the formation rate of the 4'-hydroxymethyl metabolite (M4), the basic incubation medium contained 0.1 mol/L Tris-HCl buffer (pH 7.4), 1.0 mmol/L NADPH, 10 mmol/L KCl, 10 mmol/L MgCl2, 1.0 g/L microsomal protein and 5–600 µmol/L imrecoxib in a final volume of 200 µL. The mixture was incubated at 37 °C for various times (0, 5, 15, 30, or 60 min). The reactions were initiated by the addition of NADPH after 5 min preincubation and were terminated by the addition of 50 µL cold methanol. Then 20 µL internal standard (BAP 910, 20 µmol/L in methanol) and 200 µL of 20 µmol/L NH4H2PO4 buffer (pH 3) were added to the reaction mixtures. The samples were extracted with 2 µL ethyl acetate and the supernatant was evaporated under a stream of nitrogen at 40 °C. The residue was dissolved in 100 µL of the mobile phase for LC/MSn (liquid chromatography-ion trap mass spectrometry) assay. Controls were prepared in the same manner, except for the presence of NADPH. Blank samples were assayed without substrate to exclude analytical interference by the matrix.
Metabolite identification To identify the in vitro phase I metabolites of imrecoxib formed from rat liver microsomes, 200 µmol/L of imrecoxib (saturated concentration) was used as substrate. The final volume was 1 mL and the mixtures were incubated at 37 °C for 60 min. The reactions were initiated by addition of NADPH and were terminated with 1 mL of 20 µmol/L cold NH4H2PO4 buffer (pH 3). Then the mixtures were applied to preconditioned 2.5 mL C18 cartridges (Tianjin Fuji Co, China). The columns were washed with 2 mL water and the metabolites were eluted with 1 mL methanol. The eluting solvents were evaporated and the residue was dissolved in 100 µL of the mobile phase for LC/MSn analysis.
Enzyme kinetics Linear conditions for the formation of M4 were established with respect to protein content and incubation time. The rate of formation was linear over 60 min incubation and 0.5 to 1.5 g/L of microsomal protein. The Michaelis-Menten kinetics of imrecoxib 4'-methyl hydroxylation by rat liver microsomes was determined by using 14 substrate concentrations in the range of 5 to 600 µmol/L at 37 °C for 15 min. The rate of imrecoxib metabolism was analyzed by using the MULTI program, using a nonlinear least-squares method[20]. Data were also analyzed by linear transformation (Eadie-Hofstee plot) to confirm a single Km model. The following Michaelis-Menten equation was used to analyze the relation between velocity and substrate concentra-tion:
V=Vm·S/(Km+S),
where V, S, Km, and Vm are the velocity of metabolite formation, the substrate concentration, the apparent Michaelis-Menten constant, and the maximum velocity of metabolism, respectively.
Inhibition study of imrecoxib metabolism Inhibitory effects on the 4'-methyl hydroxylation of imrecoxib in rat liver microsomes prepared from induced rats and control rats were determined at substrate concentrations of 50 µmol/L imrecoxib (~Km) and at 3 inhibitor concentrations in the range of 2–50 µmol/L. Incubations were performed at 37 °C for 15 min. The selective P450 inhibitors α-naphthoflavone (CYP 1A), quinine (CYP 2D), methylpyrazole (CYP 2E), cimetidine (CYP 2C), and ketoconazole (CYP 3A) were used. Because all the inhibitors were dissolved in methanol, an equivalent volume of methanol (without inhibitors) was included in the control incubations to correct for any effects of the solvent on microsomal activity. The concentration of methanol was 0.5%.
LC/MSn analysis A Finnigan LCQ liquid chromatography-ion trap mass spectrometer (San Jose, USA) was used to identify the in vitro metabolites of imrecoxib formed by rat liver microsomes. The instrument was operated in positive electrospray ionization mode. The source voltage was held at 4.5 kV. The capillary voltage was fixed at 13 V, and its temperature was set at 200 °C. Nitrogen was used as the sheath gas (0.75 L/min) and auxiliary gas (0.15 L/min). The MS2 spectra were produced by collision-induced dissociation (CID) of the selected precursor ions with helium in the mass analyzer, and the relative collision energies were individually optimized for each compound. Liquid chromatography was performed with a Shimadzu LC-10 AD solvent delivery system (Kyoto, Japan). The samples were separated on a Diamonsil C18 column (200 mm×4.6 mm ID; 5 µm, Dikma Technologies, Beijing, China). A mobile phase consisting of methanol-ammonium acetate 10 mmol/L (60:40, v/v) was used at a flow rate of 0.5 mL/min. The injection volume was 20 µL. All data were analyzed by using Xcalibur software (version 1.2, Thermo Finnigan MAT).
The formation rate of M4 was determined by using the same LC/MSn system, except that a mobile phase composed of acetonitrile-water-formic acid (75:25:0.5, v/v/v) was used and quantification was performed using selected reaction monitoring (SRM) of the transitions m/z 386 → m/z 356, 278 for M4 and m/z 374 → m/z 278 for BAP 910 (IS). Calibration standards were prepared by spiking 20 µL of appropriate standard solution of M4 into 200 µL of blank medium. The linear regressions of the peak area ratios versus concentrations were fitted over the concentration range of 0.10–50.0 µmol/L. The method was validated by determining quality control (QC) samples at 3 concentration levels on 3 consecutive days. The precisions were expressed as relative standard deviation (RSD) and the accuracy as relative error (RE%). The intra- and inter-day precision values were less than 8%. The accuracy for M4 was 7.5%, -5.3% and -4.8% at 0.1 µmol/L, 4.0 µmol/L and 50.0 µmol/L levels, respectively.
Codeine O-demethylase activity assay Codeine O-demethylation was carried out as a probe assay for CYP 2D[21,22]. The substrate concentration of codeine was 20 µmol/L. Incubations were performed at 37 °C for 20 min. After the reactions were finished, 40 µL internal standard (diphenhydramine, 20 µmol/L in methanol) and 200 µL of 0.1 mol/L Na2CO3 were added to the mixtures. The samples were extracted with 3 mL diethyl ether and the supernatant was evaporated under a stream of nitrogen at 40 °C. The residue was dissolved in 100 µL of the mobile phase. An aliquot of 20 µL of the solution was analyzed by using the same LC/MSn system as described for LC/MSn analysis except that the source voltage was held at 4.25 kV and its temperature was set at 180 °C; the SRM of the transitions of m/z 286 → m/z 201, 229 for morphine and m/z 256 → m/z 167 for diphenhydramine (IS) were used for quantification; and the mobile phase consisted of methanol-water-formic acid (60:40:0.5, v/v/v).
Nifedipine dehydrogenase activity assay Nifedipine dehydrogenation was used to probe the activity of CYP 3A[23]. Nifedipine (80 µmol/L) was incubated with microsomal protein at 37 °C for 10 min in a final volume of 200 µL. After the reactions were finished, 20 µL internal standard (gliclazide, 400 µmol/L in methanol) and 100 µL of 0.1 mol/L NaOH were added to the mixtures, respectively. The samples were extracted with 2 mL n-hexane-dichloromethane-iso-propyl alcohol (20:10:1, v/v/v). The dehydronifedipine formed was analyzed using the same LC/MSn system as described earlier except that the SRM of the transitions of m/z 345 → m/z 284 for dehydronifedipine and m/z 324 → m/z 127, 168 for gliclazide (IS) were used for quantification; and the mobile phase consisted of acetonitrile-water-formic acid (85:15:0.5, v/v/v).
Data analysis All data are the means of 3 individual incubations. The significance of differences between means were evaluated by using ANOVA followed by the one-tailed Student’s t-test.
Results
Metabolism of imrecoxib in rat liver microsomes Compared with the controls, 3 metabolites were found in rat liver microsomal incubates, in addition to the substrate imrecoxib (Figure 2). The structures of the metabolites were identified by investigation of their chromatographic behavior, and electrospray ionization MS and MS2 spectra relative to reference substances. The retention times and main characteristic ions in mass spectra of imrecoxib and its metabolites are summarized in Table 1.


Full table
The compound eluting at 30.2 min had the same pseudomolecular ion ([M+H]+), full scan MS2 spectrum, and chromatographic behavior as imrecoxib, therefore, it was identified as unchanged imrecoxib. By using the same method, the metabolites eluting at 8.0 min, 10.5 min, and 29.3 min were identified as the 4'-hydroxymethyl-5-hydroxyl metabolite (M3), the 4'-hydroxymethyl metabolite (M4), and the 4'-hydroxymethyl-5-carbonyl (M5) metabolite, respectively. No metabolites were detected in the absence of NADPH, indicating that metabolite formation is enzymatic and NADPH-dependent.
Enzyme kinetics Overall, the formation of M4 conformed to saturable kinetics and a representative Michaelis-Menten plot is shown in Figure 3. The Eadie-Hofstee plot (Figure 3, inset) for the formation of M4 from imrecoxib was indicative of monophasic behavior. Accordingly, a simple Michaelis-Menten kinetic analysis was used to estimate Km and Vm (Table 2). The Vm of M4 in dexamethasone-induced micro-somes increased significantly, to 7.5-fold higher than that in control microsomes. The same parameter in isoniazid-induced microsomes was 2-fold higher than that in control microsomes from rats treated with saline. The results suggest that CYP 3A and CYP 2E enzymes play important roles in the 4'-methyl hydroxylation of imrecoxib in rat liver microsomes.


Full table
Effects of inducers on the formation of M4 Imrecoxib (50 µmol/L or 200 µmol/L) was incubated at 37 °C for 15 min with induced and control rat liver microsomes (1.0 g/L) in the presence of NADPH. Among typical P450 inducers administered intraperitoneally, dexamethasone caused the most induction of M4 formation, followed by isoniazid (Table 3). The formation rates of M4 in dexamethasone-induced microsomes were 3.4-fold and 6.1-fold higher than those in control microsomes from rats treated with corn oil, respec-tively, when substrate concentrations of 50 µmol/L and 200 µmol/L were used, respectively. The corresponding values in isoniazid-induced microsomes were 1.2-fold and 1.4-fold higher than those in control microsomes from rats treated with saline, respectively. However, β-naphthoflavone had no significant effect on the metabolism of imrecoxib. The results were in accordance with those regarding the kinetics of imrecoxib 4'-methyl hydroxylation (Table 2).

Full table
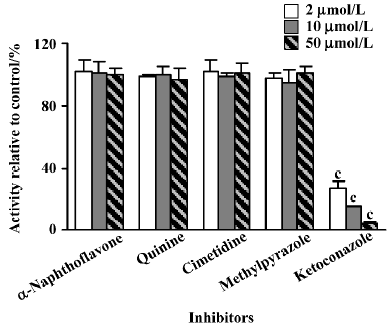
Effects of inhibitors on the formation of M4 The effects of inhibitors on the formation of M4 in rat liver microsomes are shown in Figures 4–6. The inhibitory activities are expressed as a ratio in comparison with the control activity without inhibitors. Ketoconazole, a selective inhibitor of CYP 3A, was shown to effectively decrease the formation rate of M4. When 2 µmol/L of ketoconazole was used, the hydroxylation activity was reduced to approximately 70% of the control activity in microsomes from control rats treated with saline or corn oil (Figure 4), whereas much lower enzyme activity (28% of the control activity) was observed in microsomes from dexamethasone-induced rats at the same ketoconazole concentration (Figure 5). In addition, a significant inhibition was observed in the presence of quinine (CYP 2D-selective) in control microsomes, but the other chemical inhibitors, α-naphthoflavone, cimetidine, and methylpyrazole, did not produce any significant effects on the 4'-methyl hydroxylation of imrecoxib (Figure 4). The effects of inhibitors on the formation of M4 by liver microsomes from isoniazid-induced rats and from β-naphthoflavone-induced rats were similar to those observed in control microsomes (Figure 6).


Discussion
Three phase I metabolites of imrecoxib were observed in rat liver microsomal incubates using the LC/MSn method with a saturated substrate concentration (200 µmol/L imrecoxib), a longer incubation time (60 min), and a larger final volume (1 mL). The identities of the metabolites were confirmed by chromatographic and mass spectra comparison with reference substances. These metabolites were identified as the 4'-hydroxymethyl-5-hydroxyl metabolite (M3), the 4'-hydroxymethyl metabolite (M4), and the 4'-hydroxymethyl-5-carbonyl (M5) metabolite. The formation of these metabolites was enzymatic and NADPH-dependent. However, when 50 µmol/L imrecoxib was incubated at 37 °C for 60 min in a final volume of 200 µL, imrecoxib metabolites other than M4 were not detected. Under these conditions, approximately 40% of imrecoxib was converted to M4 and the remainder was unchanged imrecoxib. It seems that 4'-methyl hydroxylation is the major metabolic pathway in NADPH-fortified rat liver microsomes. Although further oxidation of the 4'-hydroxymethyl metabolite to form the 4'-carboxylic acid metabolite (M2) was the predominant pathway in vivo, no carboxylic acid metabolite was found in the present study. This suggests that the generation of M2 in vitro may require the presence of cytosolic enzymes as in the metabolism of celecoxib[24]. This hypothesis is under investigation.
To identify the CYP isozymes involved in the 4'-methyl hydroxylation of imrecoxib, the effects of specific inducers and inhibitors of CYP on this reaction were examined. M4 was produced to the greatest extent by microsomes from dexamethasone-induced rats (Table 2 and 3). Dexamethasone is considered to be a specific inducer of CYP 3A[25,26]. However, ketoconazole, a well-known inhibitor of CYP 3A[22,26], strongly inhibited the reaction in a concentration-dependent manner (Figure 4–6). These results indicate that CYP 3A is the principal enzyme involved in the 4'-methyl hydroxylation of imrecoxib in rat liver microsomes.
In addition, 10 mmol/L of quinine (an inhibitor specific for CYP 2D[22,27]) significantly inhibited the rate of formation of M4 by approximately 20% compared with control activities in all types of microsomes except the microsomes obtained from dexamethasone-pretreated rats (Figure 4–6). When the concentration of quinine was increased to 50 µmol/L, the formation rate was decreased to a much lower level. With respect to the results mentioned above, participation of CYP 2D in the 4'-methyl hydroxylation of imrecoxib is also thought to be involved in control rats, and in β-naphthoflavone-induced and in isoniazid-induced rats.
α-Naphthoflavone, which is often used as inhibitor of CYP 1A[22,26,28], did not decrease the rate of hydroxylation of imrecoxib. This result is in agreement with those of the induction experiments. β-naphthoflavone, an inducer of CYP 1A, did not significantly increase the formation of M4 (Tables 2 and 3). Cimetidine was used to confirm the participation of CYP 2C[27,28] in imrecoxib biotransformation; however, the formation of M4 was not inhibited by cimetidine in microsomes either from control rats or from induced rats. Thus, substantial participation of CYP 2C was unlikely in this reaction. Methylpyrazole, a selective inhibitor of CYP 2E[26], had no significant inhibitory effect on the 4'-methyl hydroxylation of imrecoxib. However, the reaction was elevated significantly in microsomes from isoniazid-pretreated rats compared with those from control rats treated with saline (Table 3). Isoniazid is an inducer of CYP 2E[25]. There appears to be a discrepancy between these two different phenomena. The results of inhibition studies clearly show that CYP 3A and 2D catalyze the 4'-methyl hydroxylation of imrecoxib, so the effects of isoniazid on CYP 3A and 2D were investigated in the present study. Codeine O-demethylation and nifedipine dehydrogenation were carried out as probe assays for CYP 2D and 3A, respectively, and their activities were determined as described in the materials and methods section. The activities of codeine O-demethylase in control microsomes from rats treated with saline and in isoniazid-induced microsomes were 45.8±5.9 pmol·min-1·mg-1 protein and 59.4±7.5 pmol·min-1·mg-1 protein, respectively. The activities of nifedipine dehydrogenase in control microsomes from rats treated with saline and in isoniazid-induced microsomes were 759.8±35.4 pmol·min-1·mg-1 protein and 722.6±20.8 pmol·min-1·mg-1 protein, respectively. There was no significant difference in the activities of CYP 3A between saline-treated control microsomes and isoniazid-induced microsomes, but the activity of CYP 2D in isoniazid-induced microsomes was significantly higher than that in saline-treated control microsomes. The results suggest that the faster formation rate of M4 observed in isoniazid-induced microsomes was caused by the higher activity of CYP 2D in isoniazid-induced microsomes. So the involvement of CYP 2E in the 4'-methyl hydroxylation of imrecoxib may be excluded.
Interestingly, despite CYP 3A and 2D being shown to catalyze the 4'-methyl hydroxylation of imrecoxib, the data obtained from rat liver microsomes were described by a single Km model (Figure 3). This finding indicates that CYP 3A and 2D are likely to be characterized by similar apparent Km values.
In conclusion, imrecoxib was metabolized to 3 metabolites by rat liver microsomes: 4'-hydroxymethyl imrecoxib (M4), 4'-hydroxymethyl-5-hydoxyl imrecoxib (M3), and 4'-hydroxymethyl-5-carbonyl imrecoxib (M5). This biotransformation study of imrecoxib in rat liver microsomes indicates that 4'-methyl hydroxylation represents the major metabolic pathway of imrecoxib. The reaction was mainly catalyzed by CYP 3A. CYP2D also played a role in control rats based on the results of inhibition studies. Other CYP (such as CYP 1A, 2C, and 2E) seem to not participate in the metabolic pathway.
Acknowledgements
We would like to thank Dr Xue-qing LI (Drug Metabolism and Pharmacokinetics and Bioanalytical Chemistry, AstraZeneca Research and Development, Mölndal, Sweden) and Prof Shu-qiu ZHANG (Department of Pharmaceutics, Shanxi Medical University, Shanxi, China) for their advice and assistance.
References
- Donnelly MT, Hawkey CJ. COX-II inhibitors, a new generation of safer NSAIDS? Aliment Pharmacol Ther 1997;11:227-36.
- Jouzeau JY, Terlain B, Abid A, Nedelec E, Netter P. Cyclooxy-genase isoenzymes: how recent findings affect thinking about nonsteroidal anti-inflammatory drugs. Drugs 1997;53:563-82.
- Smith JM, Willis AL. Aspirin selectivity inhibits prostaglandin production in human platelets. Nat New Biol 1971;231:235-7.
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 1971;231:232-5.
- Riendeau D, Charleson S, Cromlish W, Mancini JA, Wong E, Guay J. Comparison of the cyclooxygenase-1 inhibitory properties of nonsteroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors, using sensitive microsomal and platelet assays. Can J Physiol Pharmacol 1997;75:1088-95.
- Masferrer JL, Zweifel BS, Manning PT, Hauser DS, Leahy KM, Smith WG, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is anti-inflammatory and nonulcerogenic. Proc Natl Acad Sci USA 1994;91:3228-32.
- Needleman P, Isakson PC. The discovery and function of COX-2. J Rheumatol 1997;24:9-14.
- Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol 1996;62:167-215.
- Lane NE. Pain management in osteoarthritis: the role of COX-2 inhibitors. J Rheumatol 1997;24:20-4.
- Simon LS, Lanza FL, Lipsky PE, Hubbard RC, Talwalker S, Schwartz BD, et al. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor: Efficacy and safety in two placebo-controlled trials in osteoarthritis and rheumatoid arthritis, and studies of gastrointestinal and platelet effects. Arthritis Rheum 1998;41:1591-602.
- Davies NM, Jamali F. COX-2 selective inhibitors cardiac toxicity: getting to the heart of the matter. J Pharm Pharm Sci 2004;7:332-6.
- Dogne JM, Supuran CT, Pratico D. Adverse cardiovascular effects of the coxibs. J Med Chem 2005;48:2251-7.
- Thomas LG. Increased risk of cardiovascular events with coxibs and NSAIDs. Lancet 2005;365:1538-9.
- Bai AP, Guo ZR, Hu WH, Shen F, Cheng GF. Design, synthesis and in vitro evaluation of a new class of novel cyclooxygenase-2 inhibitors: 3,4-diaryl-3-pyrrolin-2-ones. Chin Chem Lett 2001;12:775-8.
- Shen F, Bai AP, Guo ZR, Cheng GF. Inhibitory effect of 3,4-diaryl-3-pyrrolin-2-one derivatives on cyclooxygenase 1 and 2 in murine peritoneal macrophages. Acta Pharmacol Sin 2002;23:762-8.
- Chen XH, Bai JY, Shen F, Bai AP, Guo ZR, Cheng GF. Imrecoxib: a novel and selective cyclooxygenase 2 inhibitor with anti-inflammatory effect. Acta Pharmacol Sin 2004;25:927-31.
- Zhong DF, Zhang SQ, Sun L, Zhao XY. Metabolism of roxithro-mycin in phenobarbital-treated rat liver microsomes. Acta Pharmacol Sin 2002;23:455-60.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-75.
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 1964;239:2370-8.
- Yanaoka K, Tanigawara Y, Nakagawa T, Iga T. A pharmacokinetic analysis program (MULTI) for microcomputer. J Pharmaco-biodyn 1981;4:879-85.
- Boobis AR, Sesardic D, Murray BP, Edwards RJ, Singleton AM, Rich KJ, et al. Species variation in the response of the cytochrome P450-dependent monooxygenase system to inducers and inhibitors. Xenobiotica 1990;20:1139-61.
- Paul LD, Springer D, Staack RF, Kraemer T, Maurer HH. Cytoxhrome P450 isoenzymes involved in rat liver microsomal metabolism of californine and protopine. Eur J Pharmcol 2004;485:69-79.
- Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, et al. The conduct of in vitro and in vivo drug-drug interaction studies: a pharmaceutical research and manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003;31:815-32.
- Sandberg M, Yasar Ü, Strömberg P, Höög JO, Eliasson E. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol 2002;54:423-9.
- Yamazaki H. Rat cytochrome P450 1A and 3A enzymes involved in bioactivation of tegafur to 5-fluorouracil and auto-induced by tegafur in liver microsomes. Drug Metab Dispos 2001;29:794-7.
- Szotáková B, Skálová L, Baliharová V, Dvorščáková M, Štorkánová L, Šišpera L, et al. Characterization of enzymes res-ponsible for biotransformation of the new antileukotrienic drug quinlukast in rat liver microsomes and in primary cultures of rat hepatocytes. J Pharm Pharmacol 2004;56:205-12.
- Shimizu M, Matsushita R, Matsumoto Y, Fukuoka M. 4'-Hydroxylation of flurbiprofen by rat liver microsomes in fasting and feeding conditions. Biol Pharm Bull 2003;26:1448-54.
- Szakács T, Veres Z, Vereczkey L. Effect of phenobarbital and spironolactone treatment on the oxidative metabolism of antipyrine by rat liver microsomes. Pol J Pharmacol 2001;53:11-9.