
Pharmacokinetics of CPU0213, a novel endothelin receptor antagonist, after intravenous administration in mice1
Introduction
Endothelin (ET), a 21 amino acid peptide, is the most potent known constrictor of human resistance and capacitance vessels. It is produced in many different tissues from vascular endothelium[1], and in higher amounts from diseased myocardium, and vascular smooth muscle cells. ET-1 appears to play an important role in pathological processes underlying several cardiovascular diseases, including heart failure[2], systemic hypertension[3], myocardial infarction, atherosclero-sis, pulmonary hypertension, renal failure[4], and cardiovascular complications of diabetes, etc ET levels are elevated in both tissue and plasma[5] in various disease states. Endothelin receptor antagonists (ETRA) are currently being investigated and developed for a wide spectrum of indications[6]. Bosen-tan, the first endothelin receptor antagonist, was launched in November 2001 in the US for the treatment of pulmonary hypertension.
CPU0213 (Figure 1), synthesized by the Center of New Drug Research and Development, China Pharmaceutical University, is a new non-selective endothelin receptor antagonist that has been shown to be an effective suppressor of vasoconstriction and ET-1 binding to rat myocardial membrane preparations[7]. The pA2 values for the suppression of ET-1-induced contraction of rat thoracic aorta (mediated by ETA) and sarafotoxin (S6c)-induced contraction of guinea pig bronchial smooth muscles (ETB mediated) are 8.52±0.26 and 7.28±0.04, respectively[7], approximately 20 times the potency of bosentan[8]. CPU0213 attenuates cardiac hypertrophy[9] and chronic heart failure[10] in rats. It inhibits oxidative stress, the overexpression of matrix metalloproteinases (MMPs) and modulates the collagen content in hypertrophic myocardium induced by levothyroxine[11,12]. In hypoxia and monocrotaline (MCT)-induced pulmonary artery hypertension (PAH) model, CPU0213 markedly relieves pulmonary hypertension and right ventricular hypertrophy. The therapeutic effects of CPU0213 on vascular endothelium and kidney injury in streptozotocin (STZ)-induced diabetic rats are also notable. Therefore, it is important to investigate the pharmacokinetic behavior and obtain basic information about the pharmacokinetic characteristics of the compound. The aim of the present study is to investigate the pharmacokinetics of CPU0213 after a single iv bolus of 3 different doses of CPU0213 in relation to the acute toxic doses by iv in mice.

Materials and methods
Chemicals and reagents CPU0213 (batch N
Animals Kunming mice (equal numbers of males and females), weighing 18–22 g, were supplied by the Nanjing Jiangpu Animal Center (certificate N
Sample preparation After 5 mL of dichloromethane was added to 0.1 mL serum mixed with 0.1 mL of the internal standard (10 mg/L CPU0204), the samples were vortexed for 1 min and centrifuged at 3000×g for 10 min. The organic layer was transferred into clean tubes and evaporated at 55 ºC under a gentle stream of nitrogen. Residues were dissolved in 100 μL of the mobile phase and 20 μL was injected into HPLC column.
Chromatography The HPLC system consisted of a LC-10AT VP liquid chromatograph (Shimadzu, Japan), an SPD-10A Detector (Shimadzu, Japan). The column was a Hypersil C18, with 5 μm particle size, and 15 cm×4.6 mm internal diameter. The mobile phase comprised methanol and water (68:32, v/v). The chromatography assays were performed at 25 °C with a flow rate of 1 mL/min and UV detection at a wavelength of 254 nm.
Blank serum was assessed to identify any endogenous materials in the serum interfered with peaks of CPU0213 and an internal reference compound CPU0204.
Calibration curves for the assay of CPU0213 in serum were prepared by adding various amounts of CPU0213 to blank serum over the range of 2–560 mg/L (at 2, 4, 10, 20, 40, 80, 160, 320, 560 mg/L). All samples for the calibration curves were analyzed in the same manner as the plasma samples from dosed animals. The peak-area ratios of CPU0213 to the internal standard were calculated. Linear regression was carried out based on the peak-area ratios of CPU0213 to the internal standard plotted against the concentrations of CPU0213 spiked in serum.
Samples of CPU0213 (2, 40, 560 mg/L) were added to blank serum and assessed to determine the accuracy of the method according to the formula:
Recovery (%)=concentration prepared/concentration measured×100%.
Various concentrations of CPU0213 (2, 40, 560 mg/L) were added to blank serum and the concentration of each sample was determined 5 times to arrive at an intra-day coefficient of variation (CV) during the same day and an inter-day coefficient of variation on 5 separate days [CV (%)=SD/mean× 100%].
Pharmacokinetics assay Mice were assigned to receive CPU0213 at concentrations of 25, 50, or 100 mg/kg iv via the tail vein. At different time points (1, 5, 10, 20, 40, 60, 90, 120, 180, 240, 300, 420, and 600 min after intravenous administra-tion), blood samples were collected from the postorbital vein plexus and centrifuged. Three male and 3 female mice were used per sampling time point in each dose group. The separated serum samples were stored at -20 °C until assay. The concentrations vs time were analyzed by using the 3P87 program (version 1.0; Chinese Society of Mathematical Pharmaco-logy, Beijing, China) to calculate the pharmacokinetic para-meters.
Acute toxicity The mice were divided into 4 groups of 5 males and 5 females at random, and CPU0213 was administered iv via the tail vein at doses of 275.4, 306.0, 340.0, and 377.8 mg/kg, which were carefully determined after a series of preliminary trials. Changes in the appearance and behavior, and mortality in mice were monitored for 14 d after administration. LD50 values and the corresponding confidence limits were determined by the Bliss method[13] using NDST software (version 21; Chinese Society of Mathematical Pharmacology, Beijing, China).
Results
Chromatography validation In the present study, a sensitive and selective HPLC method was developed for the quantitative determination of CPU0213 in mouse serum. The retention times of CPU0213 and CPU0204 were 5.6 min and 3.1 min, respectively. There was no interference at these peaks, so these were free from endogenous interference (Figure 2).

The calibration curve of the peak-area ratio (y) against the respective sample concentrations (x) was constructed. The equation of mean linear regression (n=6) was y=0.0496x+0.0925 (r=0.998) for the concentration range from 2 to 560 mg/L.
The range of recovery in serum at concentrations of 2, 40, and 560 mg/L was 94.2%–103.0% (Table 1).
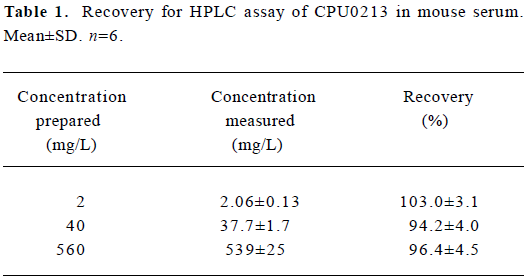
Full table
Precision was determined by 5 assays of samplings at 2, 40, and 560 mg/L. Satisfactory results were obtained for both intra-day and inter-day assays. The intra- and inter-day CV were in the range of 5.1%–5.4% and 4.9%–6.8%, respectively (Table 2).

Full table
Pharmacokinetics The serum CPU0213 concentration-time data were well characterized by a 2-compartment model (Figure 3). The initial concentrations (C0) for the 3 doses were 0.133, 0.283, and 0.581 g/L respectively. The distribution half-lives (T1/2α) and elimination half-lives (T1/2β) ranged from 1.1 to 4.2 min, and from 39.4 to 70.3 min, respectively. Parameters calculated according to statistical moment were also in the table. AUC0–t is the area under the curve from time zero to the last time point of measurable concentration. AUC0–∞ is the area under the curve from time zero to infinity. Mean residence time (MRT) was also calculated. The system serum clearance (CL) ranged from 0.00366 to 0.00544 L·min-1·kg-1 (Table 3).


Full table
Acute toxicity by iv After iv administration of the high doses of CPU0213, the mice became immediately eclamptic and their pupillae were enlarged. After 2 or 3 h, the spontaneous movements decreased and the activated muscular activity subsided. Most deaths occurred 5 min to 2 h after iv administration, and no death occurred 48h after iv administration. The mortality of mice in the 14 d after intravenous administration of CPU0213 at concentrations of 275.4, 306.0, 340.0, and 377.8 mg/kg was 0%, 50%, 70%, and 100%, respectively. The intravenous LD50 was 315.5 mg/kg and the 95% confidence limits were 301.5–330.2 mg/kg .
Discussion
Only one blood sample was obtained from each mouse. Therefore, the pharmacokinetic parameters in mice are based on mean concentration-time data. Compartment models were chosen on the basis of the Akaike Information Criterion (AIC)[14]. The R2 values of the 3 dose groups for the 2-compartment model are more than 0.999, which indicates that the concentration-time data are well fitted to a 2-compartment model. Parameters calculated by statistical moment are in agreement with those calculated by using the compartment model, providing further evidence that the 2-compartment model of the pharmacokinetic behavior is reliable.
After intravenous administration, there was a linear increase in C0 proportional to dose (r>0.9999), the same as for the AUC0–t (r=0.992) and AUC0–∞ (r=0.989). There was no prolongation of the T1/2β found with the large dose, and the CL of the 3 doses were similar. It is reasonable to surmise that CPU0213 adheres to first order rate pharmacokinetics, and no saturation was found at concentrations from 25 to 100 mg/kg.
CPU0213 is well tolerated in mice after iv bolus administration, and the acute toxicity is mild according to the LD50. The iv doses used in this pharmacokinetic study were 25, 50, and 100 mg/kg, which covers approximately one third of the LD50.
In the present study, we found that the T1/2β was not sufficiently long, being approximately 1 h following iv bolus administration. It is doubtful that CPU0213 administration once per day could offer therapeutic effects for the treatment of certain disorders. However, CPU0213 has been administered orally in other experiments, including treating animal models of pulmonary hypertension and cardiovascular complications of diabetes and hyperthyroidism[9–12]. The outcomes of these experiments show that daily oral administration at doses of 25, 50, and 100 mg/kg is sufficient to prevent pathological progression of these conditions. These data on CPU0213 provide convincing evidence that oral administration once per day provides satisfactory therapeutic efficacy, despite the short T1/2β.
The initiation of a pharmacological effect is dependent on the existence of a sufficient concentration of a drug in the plasma, and occurs when the moiety of drug is bound to the receptor site. The disappearance of a drug’s effect results from the dissociation of the drug from its receptor. In general, the effective plasma level is the dominant factor controlling a drug’s effect. The duration of drug effects, however, are not plasma level-dependent. As shown by a well-known example of a “hit and run” drug[15], even if plasma levels are negligible for a long period of time, the drug effect remains unless new enzymes are synthesized. Anticancer drugs are another group of drugs for which initiation is dependent on the effective concentration, but the duration of drug effect is not correlated with the plasma concentration of the drugs. We previously discovered that the duration of the pharmacological effect of bepridil is longer than that of the plasma concentrations of the drug[16]. This suggests that despite the T1/2β of CPU0213 being short, effective therapy can still be achieved with a single daily oral dose. It is possible that a counter-clockwise hysteresis loop[17,18] is involved in the effect–plasma level relationship, which would explain the prolonged effect against a relatively short half-life.
In conclusion, the first order rate pharmacokinetics and mild acute toxicity were observed for CPU0213. The drug effect is likely to be sustained after disappearance of the drug in the plasma, and this property should be investigated further.
References
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988;33:411-5.
- Miyuachi T, Goto K. Heart failure and endothelin receptor antagonists. Trends Pharmacol Sci 1999;20:210-7.
- Schiffrin EL. Role of endothelin-1 in hypertension. Hypertension 1999;34:876-81.
- Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension 2001;37:490-6.
- Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000;102:2434-40.
- Ray A, Hegde LG, Chugh A, Gupta JB. Endothelin receptor antagonists: current and future perspectives. Drug Discov Today 2000;5:455-64.
- Dai DZ, Huang M, Ji M, Liu LG. Endothelin receptor antagonist activity and selective blocking the ETA and ETB of compound 0213. J China Pharm Univ 2004;35:552-7.
- Clozel M, Breu V, Gray GA, Kalina B, Loffler BM, Burri K, et al. Pharmacological characterization of Bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J Pharmacol Exp Ther 1994;270:228-35.
- Huang M, Ji M, Dai DZ. The endothelin receptor antagonist 0213 reduces L-thyroxine cardiac hypertrophy and inhibits calcium mobilization in vascular smooth muscle contraction. Drug Develop Res 2002;55:12.
- Huang M, Qi JS, Dai DZ. Long-term treatment with the novel non-peptide endothelin receptor antagonist 0213 in rats with chronic heart failure. Drug Develop Res 2002;55:24.
- Liu Q, Dai DZ. Influence of a new endothelin receptor antagonist CPU0213 on expression of MMPs mRNA and TIMPs mRNA in rats with hypertrophic myocardium induced by L-thyroxin. Chin J Clin Pharmacol Ther 2004;9:281-4. Chinese..
- Liu Q, Ji M, Dai DZ. Matrix metalloproteinases changes in rat hypertrophic myocardium induced by levothyroxine and the effect of a new endothelin receptor antagonist CPU0213. J China Pharm Univ 2004;35:150-5. Chinese..
- Bliss CI. The calculation of the dosage-mortality curve. Ann Appl Biol 1935;22:134-67.
- Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr 1974;19:716-23.
- Dai DZ, Yu F, Wang YQ, Savage A. Clinical pharmacology. Beijing: The Pharmaceutical Science Publishing House of China; 2002.
- Zhu Y, Chen DD, Su L, Dai DZ. Assay of negative inotropism of bepridil on isolated guinea pig left atrial myocardium by simulating constant rate of absorption and elimination. Acta Pharmacol Sin 1993;14:161-4. Chinese..
- Sintetos AL. Pharmacokinetics and pharmacodynamics of esmolol administered as an intravenous bolus. Clin Pharmacol Ther 1987;41:112-7.
- Paalzow LK, Paalzow GHM, Tfelf-Hansen P. Variability in bioavailability: concentration versus effect. In: Rowland L, editor. Variety in drug therapy, description, estimation and control. New York: Raven Press; 1985. p 167–84.