
Design, synthesis, antitumor evaluations and molecular modeling studies of novel 3,5-substituted indolin-2-one derivatives1
Introduction
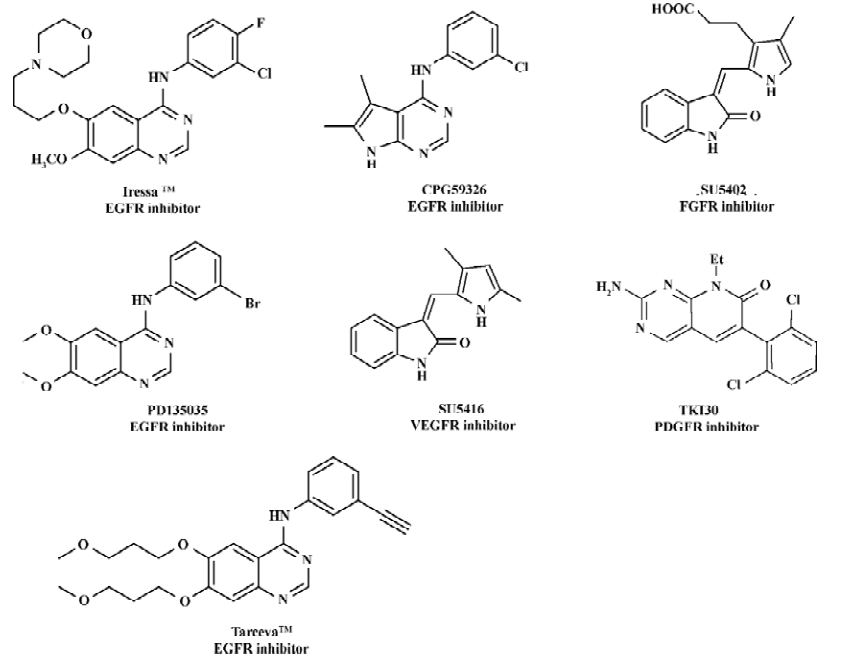
Protein tyrosine kinases (PTK) such as epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGFR), and platelet-derived growth factor receptor (PDGFR) play important roles in many of the signal transduction processes that control cell growth, differentiation, mitosis and apopto-sis[1–3]. EGFR, identified as a PTK in the 1980s, is activated by the binding of its ligands (EGF or TGF)[4] Mistakenly regulated activity or overexpression of the receptor have been demonstrated to be related to many human cancers such as breast and liver cancers[5–8], indicating that EGFR is an attractive target for antitumor drug discovery[9–13]. In recent years, a large structural variety of compounds, such as 4-anilinoquinazolines[14], 4-anilinopyrazolo[3,4-d]pyrimidines [15], 4-anilinoquinoline-3-carbonitriles[16], 7,4-anilinopyrazolo- and 4-anilinopyrrolo-quinazolines[17], were reported as EGFR tyrosine kinase inhibitors. Figure 1 includes some representative inhibitors of EGFR, VEGFR, PDGFR and FGFR which are currently approved drugs or are being clinical trial.
The indolin-2-one core is a well-known pharmacophore for developing PTK inhibitors (Figure 2). Sun et al[18,19] developed an extensive structure-activity relationship for the indolin-2-one analogs, suggesting that the inhibitory activity and selectivity of these compounds against particular PTK depends on the substituents of the indolin-2-one core, especially on the C-3 position. 3-Substituted indolin-2-ones adopting the Z configuration (R1 is substituted by pyrrol-2-yls or thien-2-yls) are potent and selective inhibitors of the FGF tyrosine kinase[18]. However, compounds with the E configuration (R1 is substituted by benzylidenyl) show fairly good potency in inhibiting EGFR tyrosine kinase[19]. The X-ray crystal structures of the FGFR tyrosine kinase in complex with 3-{[3-(2-carboxyethyl)-4-methylpyrrol-2-yl]methylene}-2-indolinone (SU5402)[20] revealed that the indolin-2-one core of SU5402 occupied the adenine pocket of the ATP binding site of the tyrosine kinase, and the substituted groups at the R1 position bound to the hydrophobic pocket of the ATP binding site (Figure 2). SU5402 with the Z configuration is a selective inhibitor of FGFR and VEGFR. What caught our attention in particular was the biological activity of E-3-substituted indolin-2-one derivatives. We introduce a β-pyrrole group into the 3-position of indolin-2-ones to see whether it would enhance the interaction between indolin-2-one compounds and tyrosine kinase and increase antitumor activity. Accordingly, a novel class of 3-pyrrole, 5-substituted indolin-2-one derivatives (1a–t) have been synthesized, and their inhibitory activities against EGFR, FGFR, VEGFR and PDGFR were determined. Significantly, compounds 1g and 1h showed promising antiproliferative effects in tumor cell lines.

Materials and methods
Chemistry
Design of analogues of compounds The inhibitory activity and selectivity of indolin-2-one analogs against particular PTK depends on the C-3 substituted groups of the indolin-2-one core. SU5402 is a selective inhibitor of FGFR and VEGFR, and bears a Z configuration. Based on the scaffold of SU5402, 20 new analogues (1a–t) (Table 1) were designed and synthesized. Keeping the key groups of SU5402, 3-substituted indolin-2-one core and pyrrole ring, we used various steric, electronic, and hydrophobic groups to substitute position 5 of the indolin-2-one core, changed the substituted position between the pyrrole ring and the indolin-2-one core, and introduced a β-pyrrole group into the 3-position of the indolin-2-ones (Table 1).

Full table
Synthetic procedures Figure 3 depicts the sequence of reactions that led to the preparation of compounds 1a–t using acetonedicarboxylates as the starting materials. In general, dimethyl-1,3-acetonedicarboxylate and diethyl-1,3-acetonedicarboxylate reacted with t-butyl acetoacetate by classic Knorr synthesis to produce 2a–b, respectively[21]. Compounds 2a–b were hydrogenated, decarboxylated and treated by Vilsmeier formylation condition, giving the key intermediates 5a–d. Indolin-2-ones were commercially available. 5-Bromoindolin-2-one (6) and 5-nitroindolin-2-one (7) were prepared by bromination and nitration of indolin-2-one[18], respectively. 5-Carboxyindolin-2-one[18] (8) was afforded by hydrolysis of 5-chloroacetylindolin-2-one, which was prepared by chloroacetylation of indolin-2-one. 5-(amino- sulfonyl) and the other 5-(substituted aminosulfonyl)-indolin-2-ones (compounds 10a–i) were obtained by amidation of 5-(chlorosulfonyl)indolin-2-one (9), which was prepared by sulfonylation of indolin-2-one with chloro-sulfonic acid. Compounds 1a–t were synthesized by condensing pyrrole aldehydes (5a–d) and 5-substituted indolin-2-ones in the presence of piperidine.

Biology assay
Tyrosine kinase assays by ELISA The tyrosine kinase assays were measured using the ELISA assay as previously described[22]. The tyrosine kinase activities of the purified EGFR, FGFR, VEGFR and PDGFR were determined in 96-well ELISA plates pre-coated with 20 µg/mL Poly(Glu, Tyr)4:1 (Sigma Chemical Co, St Louis, MO, USA). Briefly, 85µL of 8 µmol/L ATP solution diluted in kinase reaction buffer solution (50 mmol/L HEPES, pH 7.4, 20 mmol/L MgCl2, 0.1 mmol/L MnCl2, 0.2 mmol/L Na3VO4), 1 mmol/L dithiothreitol (DTT) was added to each well. Then, 10 µL of diluted compounds were added to each reaction well at varying concen-trations. 0.1% DMSO (v/v) was used as the negative control. Experiments at each concentration were performed in triplicate. The kinase reaction was initiated by the addition of purified tyrosine kinase proteins diluted in 10 µL of kinase reaction buffer solution. After incubation for 60 min at 37 °C, the plate was washed 3 times with phosphate buffered saline (PBS) containing 0.1% Tween 20 (T-PBS). Next, 100 µL of antiphosphotyrosine (PY99) antibody (1:500 dilution) diluted in T-PBS containing 5 mg/mL bovine serum albumin (BSA) was added. After 30 min incubation at 37 °C, the plate was washed 3 times. Goat anti-mouse IgG horseradish peroxidase (100 µL, 1:2000 dilution) diluted in T-PBS containing 5 mg/mL BSA was added. The plate was reincubated at 37 °C for 30 min and washed as before. Finally, 100 µL solution (0.03% H2O2, 2 mg/mL o-phenylenediamine in citrate buffer 0.1 mol/L, pH 5.5) was added and incubated at room temperature until color emerged. The reaction was terminated by the addition of 100 µL of H2SO4 (2 mol/L), and A492 was measured using a multi-well spectrophotometer (VERSAmaxTM, Charlot-tesville, VA, USA). The inhibition rate (IR; %) was calculated with Equation 1.
IR=(1–[A492/A492(control)])×100% (1)
Compounds PD135035, SU5402, SU5416 and TKI30 were used as positive controls for the EGFR, FGFR, VEGFR and PDGFR kinase, respectively.
Cell growth inhibition assay by sulforhodamine B (SRB) Three human carcinoma cell lines, A-431 (epidermoid carcinoma), A-549 (lung carcinoma) and MDA-MB-468 (breast carcinoma), obtained from American Type Culture Collection (Rockville, MD, USA), were used for the cell proliferation assay. Cells were maintained in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco, Grand Island, NY, USA) in a highly humidified atmosphere of 95% air with 5% CO2 at 37 °C. The growth inhibition was analyzed by the sulforhodamine B (SRB; Sigma, USA) assay[23]. Briefly, the cells were seeded at 6000 cells/well in 96-well plates (Falcon, Oxnard, CA, USA), and allowed to attach overnight. The cells were treated in triplicate with grade concentrations of compounds at 37 °C for 72 h. Then they were fixed with 10% trichloroacetic acid and incubated for 60 min at 4 °C. The plates were then washed and dried. SRB solution (0.4% w/v in 1% acetic acid) was added and the culture was incubated for an additional 15 min. After the plates were washed and dried, the bound stain was solubilized with Tris buffer, and the optical densities were read on the plate reader (model VERSAmax, Molecular Devices, USA) at 515 nm (A515). The growth inhibitory rate (GIR) of treated cells was calculated by Equation 1.
The results were also expressed as IC50 (the compound concentration required for 50% growth inhibition of tumor cells), which was calculated by the Logit method. The mean IC50 was determined from the results of 3 independent tests.
Cell growth inhibition assay by MTT The cancer cell line Autosomal Dominant Polycystic Kidney disease (ADPKD), which was provided by Shanghai Changzheng Hospital (Shanghai, China), was used for the cell proliferation assay. The cell was cultivated in DMEM/F12 (Gibco, USA) supplemented with 100 mL/L fetal calf serum (FCS; Sijiqing Company, Hangzhou, China), penicillin (100 µg/mL) and streptomycin (100 µg/mL) in a humidified atmosphere of 5% CO2 at 37 °C. rhEGF was obtained from Calbiochem Company (Darmstadt, Germany).
The growth inhibition was evaluated by the modified MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assay[24]. Briefly, the cells were seeded at 1×104 cells/well in 96-well plates (Falcon, USA), and incubated for 24 h in 100 mL culture media with 10% FCS. Then the media were replaced by serum-free medium. After 24 h, the media were placed in triplicate with grade concentrations of compounds, 1 ng/mL hEGF and 2% FCS medium at 37 °C for 72 h. Then the cells were treated with MTT (Sigma, USA) 10 µL (5 g/L) for 4 h. After the removal of the supernatant, the purple-blue sediment dissolved in 100 µL/well DMSO, and the optical densities were read on a multi-well scanning spectrophotometer (Labsystems Dragon, Finland) at 490 nm (A490). The GIR of the treated cells was calculated by Equation 1.
The results were also expressed as IC50 (the compound concentration required for 50% growth inhibition of tumor cells), which was fitted by using the sigmoidal fitting model by the Origin7.0 software[25]. The mean IC50 was determined from the results of 3 independent tests.
Molecular modeling The crystal structures of the kinase domain of EGFR in complex with its inhibitor Tarceva (AQ4774, PDB entry 1M17)[26] was acquired from the Brook-haven Protein Database (PDB) (http://www.pdb.org). The missing atoms and residues were reconstructed using Sybyl 6.8[27]. Kollman-united-atom charges[28],and Gasteiger-Marsili charges[29] were assigned to the protein and inhibitor, respec-tively.
To find the binding mode of SU5402 and compound 1 with EGFR, the advanced docking program AutoDock 3.0.3[30,31] was used to automatically dock the inhibitors to the binding site of EGFR. The Lamarckian genetic algorithm (LGA)[31] was applied to identify the binding orientation and conformation of these 2 inhibitors interacting with EGFR. A Solis and Wets local search performed the energy minimization on a user-specified proportion of the population. The number of generation, energy evaluation, and docking runs were set to 3.7×105, 1.5×106, and 20, respectively. The docked conformations of the inhibitor were generated after a reasonable number of evaluations. Typically, 5 binding energy terms used in current version of AutoDock 3.0[30,31] were included in the scoring function: the van der Waals interaction represented as a Lennard-Jones 12–6 dispersion/repulsion term; the hydrogen bonding represented as a directional 12–10 term; the Coulombic electrostatic potential; desolvation upon binding; and the hydrophobic effect (solvent entropy changes at solute-solvent interfaces).
The whole docking operation in this study could be stated as follows: first, the binding site of EGFR was checked for polar hydrogen and assigned for partial atomic charges, then a PDBQ file was created, and then atomic solvation parameters were assigned for the protein. Meanwhile, some of the torsion angles of the inhibitor that would be explored during the molecular docking stage were defined, allowing the conformation search for the inhibitors during the docking process. Second, the grid map with 80×80×80 points and a spacing of 0.375Å was calculated using the AutoGrid program[29] to evaluate the binding energies between the inhibitors and the protein. Third, some important parameters for LGA calculations were reasonably set, not only the atom types, but also the generations and the number of runs for the LGA algorithm were edited and properly assigned according to the requirement of the Amber force field[32,33]. The maximum number of generations, energy evaluations, and docking runs were set to 3.0×105, 1.5×106, and 30, respectively. Finally, the inhibitor-protein complex derived from docking was selected according to the criterion of interaction energy combined with geometrical matching quality.
Results
Analogue synthesis and structural characteristics In total, 20 new compounds (1a–t) were designed and synthe-sized, and their chemical structures are shown in Table 1. These compounds were synthesized through the routes outlined in Figure 3, and the details for synthetic procedures and structural characterizations are described in the Discussion section.
Only one isomer of the product was detected in thin layer chromatography (TLC) analysis and 1H NMR spectrum. The chemical shifts of the vinyl proton in documented 3-substituted indolin-2-ones were around 7.85–8.53 ppm for the Z isomer, and 7.45–7.84 ppm for the E isomer[18]. The chemical shifts of the vinyl protons of compounds 1a–t are consistent with that of the related E isomers reported[18]. The 13C-NMR chemical shifts of the β-carbon of α, β-unsaturated carbonyl compounds 1d and 1e are 147 ppm and 141 ppm, respectively.
The absolute configuration of 1a was finally confirmed by X-ray structural analysis with a yellow needle single crystal, obtained by slow evaporation of a dilute solution in EtOH/H2O (40:1). The X-ray crystal structure of 1a is shown in Figure 4A. The pyrrole ring and the carbonyl O of the indolin-2-one are at the opposite sides of the double bond, indicating that 1a is E isomeric. The superposition of the AutoDock predicting conformation of 1a with the X-ray structure is shown in Figure 4B. The root mean square deviation between these 2 conformations is ~0.386Å, and the major deviation is from the flexible moiety –CH2CO2CH2CH3, indicating that the bioactive conformation of 1a is similar to its crystal structure.

Biological activities The enzyme assay data are summarized in Table 1. Disappointingly, most compounds only displayed low to moderate inhibition activity against EGFR, FGFR, VEGFR and PDGFR at the concentration of 10 µmol/L. To some extent, compounds 1a, 1b, 1c, 1g, and 1h exhibited a better ability to inhibit the EGFR and VEGFR kinase (percent inhibition at 10 µmol/L >20.0%) than the FGFR and PDGFR kinase. However, the cellular assay turned out more encouraging (Table 2). Four human carcinoma cell lines of A-431, A-549, MDA-MB-468 and ADPKD were chosen for the cell proliferation assay. The results indicate that compounds 1d, 1g and 1h show promising anti-proliferation activities for A-431, A-549 and MDA-MB-468 (percent inhibition rates at 10 µmol/L>50%; Table 2). It is remarkable that 1d and 1e show fairly good activity against ADPKD (IC50=0.1 and 3.7 µmol/L, respectively). Compounds 1d and 1g substituted with –CH2CO2Me and methyl groups in the 3’-C position of the pyrrole ring are much more potent than compounds 1b and 1j substituted with –CH2CO2Et. The bromination or nitration at the C-5 position of the indolin-2-one core increased the potency (1d>1c, 1g>1e). The inconsistency between the enzyme activity and the cellular efficiency could imply that the new type of indolin-2-one compounds might inhibit multiple key proteins involved in the EGFR and VEGFR signaling pathways, not only targeting the tyrosine kinase, thus leading to the significant antiproliferation effect against EGFR- dependent tumor cell lines, which is highly relevant to the overexpression of EGFR or VEGFR kinase. The definite mechanism is still under study.

Full table
Molecular modeling We docked the structures of SU5402 and compound 1a into the active site of EGFR-TK by using AutoDock 3.0.3[30,31]. The predicted bioactive conformations of SU5402 and compound 1a are shown in Figure 5. The ATP binding pocket of EGFR consists of Thr766, Gln767, Leu768, Met769, Gly772, Thr830, Asp831, Val702, Lys721, Ala719, Glu738, and Met742[5]. This binding pocket can be divided into 3 regions, including 2 hydrophobic regions and an adenine region[5]. The adenine region mainly comprises Gln767 and Met769[5]. Hydrophobic region I shaped by Ala719–Lys721, Leu764–Thr766, Thr830 and Asp831 is located deep inside the ATP binding pocket[5]. Hydrophobic region II mainly comprises Leu694 and Gly772[5]. SU5402 formed 1 hydrogen bond with Thr766, and 6 hydrophobic interactions with Leu694, Val702, Ala719, Lys721, Leu830 and Asp831 (Figure 5A). The intramolecular hydrogen bonding between the N atom of the pyrrole ring and the carboxyl O atom of the indolin-2-one core in SU5402 is responsible for the Z isomeric form[18] (Figure 5A). When changing the α-pyrrole ring to a β-pyrrole ring, the distance between these 2 atoms is lengthened, and the introduction of a methyl group in the 5'-position of the pyrrole ring avoids the intramolecular hydrogen bond formation. Figure 5B shows the interaction model of compound 1a and EGFR. The indolin-2-one core of compound 1a occupies the adenine pocket of EGFR, and the pyrrole moiety lies in the hydrophobic region. Compound 1a forms 3 hydrogen bonds with Met769 and Thr830, and 8 hydrophobic interactions with Leu694, Phe699, Val702, Ala719, Ile720, Lys721, Arg817 and Leu820. This binding model is similar to that of Tarceva and EGFR[26]. The quinazoline and anilino moieties of Tarceva also occupied the adenine pocket and the hydrophobic region, respectively[28]. There are more hydrogen bonds and hydrophobic interaction pairs between 1a and EGFR than with SU5402 and EGFR.

Discussion
In summary, by replacing the α-pyrrole ring at the 3-position of the indolin-2-one core of Z-SU5402 with a β-pyrrole ring, and introducing a methyl group at the 5'-position of a pyrrole ring, we successfully designed and synthesized a series of E-3-substituted indolin-2-ones. Biological assay indicated that 4 compounds (1d, 1e, 1g, and 1h) exhibited promising inhibitory activity toward the A-549, MDA-MB-468, and ADPKD cell lines. Compounds 1g and 1h provide a promising new template for further development of antitumor agents.
References
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990;61:203-12.
- Aaronson SA. Growth factors and cancer. Science 1991;254:1146-53.
- Boschelli DH, Wu Z, Klutchko SR, Showalter HDH, Hamby JM, Lu GH, et al. Synthesis and tyrosine kinase inhibitory activity of a series of 2-amino-8H-pyrido[2,3-d]pyrimidines: identification of potent, selective platelet-derived growth factor receptor tyrosine kinase inhibitors. J Med Chem 1998;41:4365-77.
- Alexander JB. Chemical inhibitors of protein kinases. Chem Rev 2001;101:2541-72.
- Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 1999;82:195-206.
- Cance WG, Liu ET. Protein kinase in human breast cancer. Breast Cancer Res Treat 1995;35:105-14.
- Chrysogelos SA, Dickson RB. Egf receptor expression, regulation, and function in breast cancer. Breast Cancer Res Treat 1994;29:29-40.
- Yang EB, Wang DF, Cheng Y, Mack P. Expression and functions of growth-factors and growth-factor receptors in liver-cancer. Cancer J 1997;10:319-24.
- Yang EB, Guo YJ, Zhang K, Chen YZ, Mack P. Inhibition of epidermal growth factor receptor tyrosine kinase by chalcome derivatives. Biochim Biophys Acta 2001;1550:144-50.
- Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, et al. Tyrosine kinase inhibitors: from rational design to clinical trials. J Med Res Rev 2001;21:499-512.
- Kurup A, Garg R, Hansch C. Comparative QSAR study of tyrosine kinase inhibitors. Chem Rev 2001;101:2573-600.
- Khazaie K, Schirrmacher V, Lichtner RB. EGF receptor in neoplasia and metastasis. Cancer Metast Rev 1993;12:255-74.
- Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, et al. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science 1994;265:1093-5.
- Smaill JB, Rewcastle GW, Loo JA, Greis KD, Chan OH, Reyner EL, et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J Med Chem 2000;43:1380-97.
- Traxeler P, Bold G, Frei J, Lang M, Lydon N, Mett H, et al. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J Med Chem 1997;40:3601-16.
- Wissner A, Overbeek E, Reich MF, Floyd MB, Johnson BD, Mamuya N, et al. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3- carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J Med Chem 2003;46:49-63.
- Palmer BD, Trumpp-Kallmeyer S, Fry DW, Nelson JM, Showalter HDH, Denny WA. Tyrosine kinase inhibitors. 11. Soluble analogues of pyrrolo- and pyrazoloquinazolines as epidermal growth factor receptor inhibitors: synthesis, biological evaluation, and modeling of the mode of binding. J Med Chem 1997;40:1519-29.
- Sun L, Tran N, Liang C, Tang F, Rice A, Schreck R, et al. Design, synthesis, and evaluations of substituted 3-[(3-or4-carboxyethyl-pyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. J Med Chem 1999;42:5120-30.
- Sun L, Tran N, Tang F, App H, McMahon G, Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem 1998;41:2588-603.
- Mohammadi M, McMahon G. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997;276:955-60.
- Battersby AR, Hunt E, McDonald E, Paine JB III, Saunders J. Biosynthesis of porphrins and related macrocycles. Part VIII. Enzymic decarboxylation of uroporphyrinogen-III: structure of an intermediate, phyriaporphyrinogen-III, and synthesis of the corresponding porphyrin and of two isomeric porphrins. J Chem Soc Perk 1976;I:1008-21.
- Guo XN, Zhong L, Zhang XH, Zhao WM, Zhang XW, Lin LP, . Evaluation of active recombinant catalytic domain of human ErbB-2 tyrosine kinase, and suppression of activity by a naturally derived inhibitor, ZH-4B. Biochim Biophys Acta 2004; 1673: 186–93.
- Wang S, Yang D, Enyedy I. Erbb-2 selective small molecule kinase inhibitors. US patent 2004023957-A1. 2004.
- Mosmann T. An in vitro murine model of a penicillin specific IgE anamnestic response. J Immunol Methods 1983;139:55-60.
- Origin. Version 7.0. Northampton, MA: Microcal Software Inc, 2002. [cited 2005 Jul 20]. Available from: .www.microcal.com
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res 2000;28:235-42.
- Sybyl molecular modeling package (Computer program). Version 6.8. St Louis, MO: Tripos Associates, 2000.
- Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 1984;106:765-84.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity — a rapid access to atomic charges. Tetrahedron 1980;36:3219-88.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, . Autodock (Version 3.0.3), Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA, USA. 1999.
- Case DA, Pearlman DA, Caldwell JW, Cheatham TE III, Ross WS, Simmerling CL, . AMBER 5.1, 5th ed, San Francisco, CA: University of California; 1997.
- Liu H, Li Y, Song M, Tan X, Cheng F, Zheng S, et al. Structure-based discovery of potassium channel blockers from natural products: virtual screening and electrophysiological assay testing. Chem Biol 2003;10:1103-13.
- POV-ray. Version 3. POV-ray-Team, 1999. [cited 2005 Jul 20]. Available from: .www.povray.org
- Usack KP, Arnold LD, Barberis CE, Chen HP, Ericsson AM, Gaza-Bulseco GS, et al. A 13C NMR approach to categorizing potential limitations of a, b-unsaturated carbonyl systems in drug-like molecules. Bioorg Med Chem Lett 2004;14:5503-7.
- John B, Dolphin D. Pyrrole chemistry. An improved synthesis of ethyl pyrrole-2-carboxylate esters from diethyl aminomalonate. J Org Chem 1985;50:5598-604.
- Sheldrick GM. SHELXS-97: Program for the solution of crystal structure. Germany: Goettingen Univ; 1997.
- Beurskens PT, Admiraal B, Beurskens G, Bosman WP, Garcia-Granda S, Gould RO, . The DIRIDIF-94 program system. Technical report of the crystallography laboratory. The Nether-lands: University of Nijmegen, 1994.
- Cromer DT, Waber JT. International tables for X-ray crystallo-graphy. Vol 4. Kynoch Press and Birmingham: England, 1974.
- Ibers JA, Hamilton WC. Dispersion corrections and crystal structure refinements. Acta Cryst 1964;17:781-2.
- Creagh DC, Hubbell JH. International tables for crystallography. In: Wilson AJC editor. Boston: Kluwer Academic Publishers: 1992.