
CYP3A4 mediated in vitro metabolism of vinflunine in human liver microsomes1
Introduction
Vinca alkaloids, including the natural products vinblastine (VBL) and vincristine (VCR) and the semisynthetic derivatives vindesine (VND) and vinorelbine (VRL), are antimitotic drugs that are widely and successfully used in the treatment of cancer. Their interactions with tubulin, the major component of microtubules in the mitotic spindle, and the subsequent arrest of cells in mitosis are generally accepted as key events in their mechanisms of action[1,2]. Although VBL and VCR have reasonable potency in clinics, there are considerable toxic side effects associated with them. Semisynthetic development of the Vinca alkaloids has produced a successful second-generation compound, VRL, which has shown improved efficacy and reduced toxicity[3]. It is effective in the treatment of non-small cell lung cancer, metastatic breast cancer and ovarian cancer, and it has shown promise in the management of lymphomas, esophageal cancer and prostatic carcinomas[4–6]. Now, it has been further modified through super-acidic chemistry to generate new and more active derivatives. The process of production involves the insertion of 2 fluorine atoms at the 20' position and reduction of the 3'4' double bond to produce 20',20'-difluoro-3'4'-dihydrovinorelbine, known as vinflunine (Figure 1)[3].
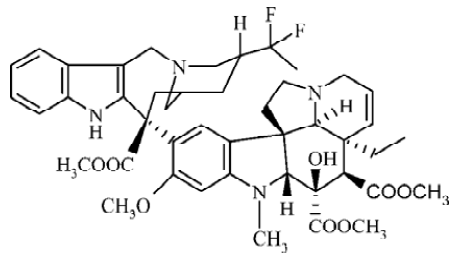
As a new vinca alkaloid, vinflunine exerted markedly superior effects on antitumor activities against a panel of 13 murine and human tumor models compared to the parent compound, VRL[7–9]. In vitro investigations have confirmed the mitotic-arresting and tubulin-interacting properties of vinflunine shared by other Vinca alkaloids[10,11]. However, differences in terms of the inhibitory effects of vinflunine on microtubules dynamics and its tubulin-binding affinities have been identified, which appear to distinguish it from the other Vinca alkaloids[12]. Vinflunine induced smaller spirals with a shorter relaxation time, which might be associated with reduced neurotoxicity[13]. Furthermore, an in vivo study has suggested that vinflunine mediates its antitumor activity at least in part via an antivascular mechanism, even at subcyto-toxic doses[14]. Although vinflunine appeared to participate in P-glycoprotein-mediated drug resistance mechanisms, it proved only a weak substrate for this protein and a far less potent inducer of resistance than VRL[15]. Vinflunine is presently in phase III experimentation for treatment of bladder cancer and non-small-cell lung cancer[16].
Vinca alkaloids are rapidly and extensively converted by human hepatocytes to a number of unidentified biotransformation products[17]. In addition, it is reported that CYP3A catalytic activities made a major contribution to the overall metabolism of VBL and VND in human liver microsomes. However, the chemical structures of these metabolites remain unknown[18,19]. It has also been reported that VRL is metabolized to 3 metabolites, 4-O-deacetylvinorelbine, 20' hydroxyvinorelbine and VRL 6'-oxide, in human blood[20]. Five metabolites, 15,16-epoxyvinorelbine, 11'-hydroxyvino-relbine, 19'-hydroxyvinorelbine, 15,16-epoxy-10'-hydro-xyvinorelbine and 10'-hydroxyvinorelbine, are identified in rats[21]. Also, it is reported that CYP3A4 is mainly responsible for the metabolism of VRL in human liver microsomes[22,23].
Compared to the extensive literatures on pharmacodynamic investigations, very little information is available on the pharmacokinetics of vinflunine. The purpose of these experiments was to investigate the metabolism of vinflunine in human liver microsomes in vitro, and to identify the major cytochrome P-450 (CYP450) isoform involved in the metabolism of vinflunine.
Materials and methods
Chemicals Vinflunine was supplied by Qilu Pharmaceutical Co, Ltd (Ji-nan, China). The internal standard VCR was a gift from Organic Chemistry Laboratories of China Pharmaceutical University (Nanjing, China). Glucose-6-phosphate dehydrogenase (G-6-PDH, Type V), α-naphthoflavone (α-Naph), sulfaphenazole (Sul), quinidine (Qui), diethyl dithiocarbamate (DDC) and tranylcypromine (Tra) were purchased from Sigma Chemical Co (St Louis, MO, USA). Ketoconazole (Ket) was kindly provided by Nanjing Second Pharmaceutical Factory. α-Nicotinamide adenine dinucleotide phosphate (NADP) and G-6-P were purchased from Shanghai Lizhu-dongfeng Biotechnological Co (Shanghai, China). All other supplies were of the highest grade available from standard commercial sources.
Tissue samples and preparation of liver microsomes The human liver used in the present study was obtained from Jinling Hospital (Nanjing, China), and the study was approved by the Ethics Committee of Jinling Hospital. The human liver microsomes were prepared by differential centrifugation[24]. The microsomal protein concentration was determined by the method of Lowry.
Incubation and sample preparation The incubation conditions of the experiment were established and controlled to provide a reproducible and linear rate of the metabolite. A typical incubation mixture consisted of potassium phosphate buffer (PH 7.4) 100 mmol/L, an NADPH generating system (MgCl2 5 mmol/L, G-6-P 10 mmol/L, NADP 1 mmol/L, G-6-PDH 1 kU/L), vinflunine 10 µmol/L, and microsomal protein 1 g/L, in a final volume of 1 mL[17]. The reaction was initiated by the addition of the NADPH generating system. After incubation at 37 °C for 1 h, the reaction was terminated by adding 200 µL of NaOH 1 mmol/L. Vinflunine and its metabolites, M1, M2, in the incubation mixture were determined by the LC mass solution method. After adding 1 µg/mL VCR as the internal standard, the reaction mixtures were extracted with 5 mL of ethyl acetate and centrifuged at 2100×g for 10 min. The organic fraction evaporated to dryness using an evaporator at 50 oC. The residue was reconstituted in 100 µL of the mobile phase. After centrifugation at 9000×g for 10 min, a 10 µL aliquot of the solution was injected into the chromatographic system.
The HPLC system consisted of a DGU-20A3 degasser (Shimadzu corporation, Kyoto, Japan), Shimadzu 20AD pumps (Shimadzu corporation, Kyoto, Japan), a high pressure mixer, a CTO-20A column oven (Shimadzu corporation, Kyoto, Japan) and a Shimadzu SIL-20AC autosampler(Shimadzu corporation, Kyoto,Japan). A Shimadzu 2010 liquid chromatograph-mass spectrometer (Shimadzu corpora-tion, Kyoto,Japan) equipped with an electrospray ionization (ESI) probe, QoQ system (Q-array-octapole-quadrupole mass analyzer; Shimadzu corporation, Kyoto, Japan) was used in the study. The analysis was carried out on an ODS column (Shim-pack, 5 µm, 2.1mm×250 mm ID; Shimadzu, Japan). The mobile phase was acetonitrile, 1 mmol/L ammonium acetate (35:65, v/v), and the column temperature was maintained at 35 oC. A constant mobile phase flow-rate of 0.2 mL/min was employed throughout the analyses. Mass spectrometric conditions were optimized to obtain maximum sensitivity. The final ESI conditions used were as follows: curve dissolution line voltage was fixed as that in Tuning and the probe high voltage was set at 4.5 kV, Q-array voltage of DC -35 V and RF 150 V. Mass spectra were obtained at a dwell time of 0.2 s in selected ion monitor (SIM) mode and 1 s in scan mode. Liquid nitrogen (99.995%, from Gas Supplier Center of Nanjing University, Nanjing, China) was used as the nebulizing gas at 4.5 L/min. LCMS Solution Version 3.2 (Shimadzu corporation, Kyoto, Japan) worked on Windows 2000. Vacuum in the mass detector was obtained by a Turbomo-lecular pump (Pfeiffer Vacuum GmbH, Asslar, Germany). Vinflunine in the incubation mixture were determined by mass analysis[25]. The metabolites were quantitated by peak area ratio.
Inhibition study The effects of various selective CYP450 inhibitors on the formation of metabolites M1 and M2 in human liver microsomes were investigated. The inhibitors studied were α-Naph (CYP1A2), Qui (CYP2D6), DDC (CYP2E), Sul (CYP2C9), Tra (CYP2C19) and Ket (CYP3A). The concentration of vinflunine was 10 µmol/L, and the concentration range of inhibitors was 2.5–20 µmol/L for Qui, 12.5–100 µmol/L for α-Naph, DDC, Tra and Sul, and 0.5–5 µmol/L for Ket.
Study of inhibition type After identifying the major metabolic enzyme of vinflunine in vitro, we incubated vinflunine in human liver microsomes in the absence and presence of Ket for 30 min, and then used a graphical method for analyzing enzyme data to identify the types of inhibition of Ket on the formation of M1 and M2. The concentration range of vinflunine was 2.5–40 µmol/L and Ket was 0.25–5 µmol/L.
Results
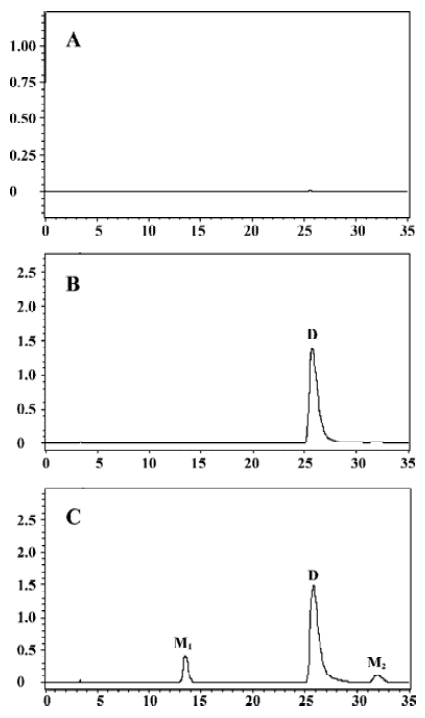
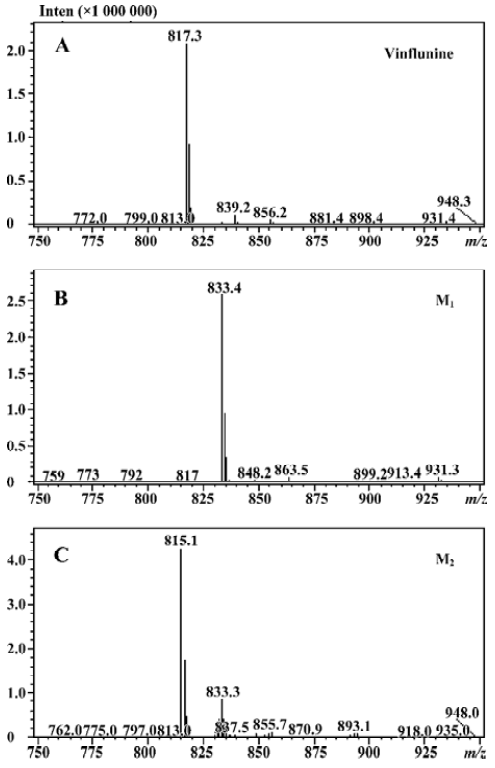
Microsomal incubation of vinflunine in the absence of the NADPH-generating system resulted in a single peak of parent vinflunine. Vinflunine metabolism by human liver microsomes was found to be an NADPH-dependent process. For the optimization of both reaction time and protein concentration, M1 and M2 formations were tested and the condition was chosen within the linear rang of these 2 reactions. M1 and M2 formed linearly with the reaction time less than 45 min and protein concentration up to 2 mg/mL. The substrate concentrations were determined based on the Km values for both M1 and M2 which were estimated by the Michaelis-Menten saturation curve in preliminary experi-ments. Following the incubation of vinflunine with human liver microsomes and the NADPH-generating system, vinflunine was rapidly metabolized, and 2 metabolites (M1, M2) were isolated in the incubation mixture (Figure 2). Direct HPLC/MS analysis using an electrospray ionization interface under positive ion mode resolved 2 drug-related compounds in the incubations (Figure 3). M1 exhibited the protonated molecular ion [M+H]+ at m/z 833, and M2 exhibited the protonated molecular ion [M+H]+ at m/z 815. Thus, the molecular weight of M1 and M2 were 832 and 814, respectively. According to mass spectra analysis and the metabolic pathway of VRL[18,19], we can tentatively presume that M1 was the N-oxide metabolite or hydroxylated metabolite, and M2 was the epoxide metabolite of vinflunine, although this needs further identification. The proposed metabolic pathway of vinflunine is presented in Figure 4.

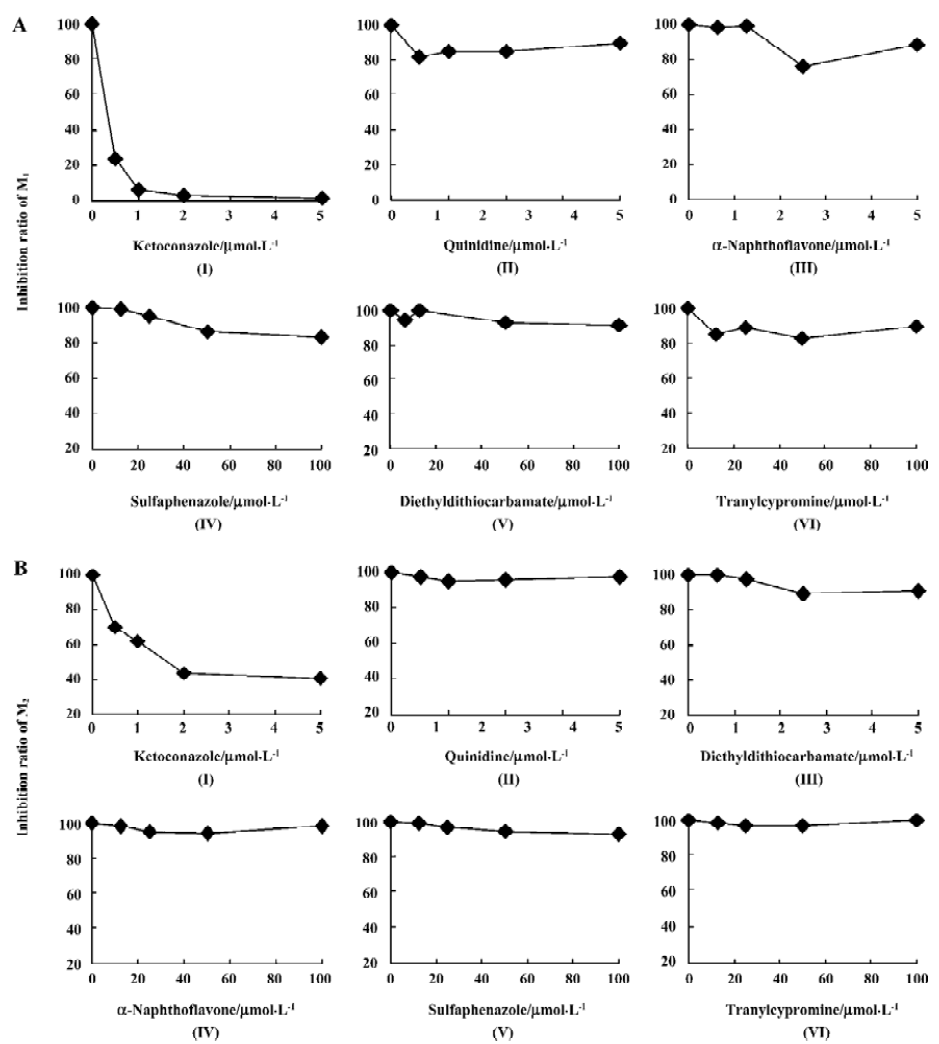
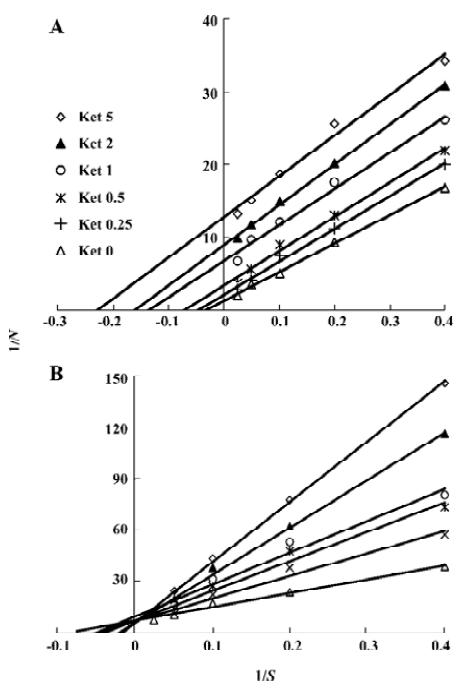
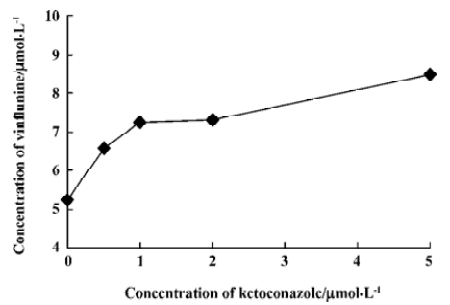
Ket, the specific inhibitor of CYP3A, could inhibit the formation of M1 and M2 while other inhibitors had no significant inhibitory effects on their formation (Figure 5). The Lineweaver-Burk plot of vinflunine metabolism activity in human liver microsomes in the absence or presence of Ket is presented in Figure 6. Ket uncompetitively inhibited the formation of M1 and competitively inhibited the formation of M2, resulting in a lower rate of vinflunine metabolism in human liver microsomes (Figure 7).
Discussion
Biotransformation of vinflunine in human liver microsomes is NADPH dependent; this suggests that liver microsome CYP450 was involved in the metabolism of vinflunine. After incubation with human microsomes, vinflunine was rapidly metabolized to 2 metabolites, M1 and M2. Direct HPLC/MS analysis using an electrospray ionization interface under positive ion mode resolved 2 drug-related compounds in the incubations. The molecular weight of M1 (832) is 16 more than that of its parent drug (816). Vinflunine, 20'-difluoro-3'4'-dihydrovinorelbine, is similar to VRL in chemical structure, while 20'-hydroxyvinorelbine and VRL N-oxide was reported to be the metabolites of VRL in humans[20]. So we can prospect that M1 was vinflunine N-oxide metabolite or hydroxylated metabolite. The molecular weight of M2 (814) is 2 less than that of vinflunine. According to the metabolic pathway of VRL in rats[21], M2 was prospected to be epoxy vinflunine.
Double reciprocal plots indicated that Ket, the specific inhibitor of CYP3A, could inhibit the formation of M1 by an uncompetitive mechanism, while M2 could inhibit formation by a competitive mechanism. The results of inhibition study suggest that CYP3A4 is a major CYP450 isoform involved in the metabolism of vinflunine, which is consistent with reports that CYP3A4 was the main CYP450 enzyme involved in the VRL metabolism[22,23]. CYP3A4 represents the major CYP450 isozyme in the human liver (about 30%)[26]. Fifty percent of marketed drugs belonging to various therapeutic areas (including antidepressants, HIV antivirals, calcium channel blockers, anticancer agents) are metabolized by the CYP3A family and especially CYP3A4. As a consequence, some drug-drug interactions might occur in clinical practice and precisely for anticancer therapy as several drugs may be in combination with vinflunine, such as docetaxel, tamoxifen, and etoposide. However, we have not yet examined the effect of vinflunine on the metabolism of those anticancer drugs. Because of the narrow therapeutic range of these drugs, further studies are needed to investigate the effect of vinflunine on the metabolism of anticancer drugs that are likely to be co-administered with vinflunine. In addition, large interindividual variations of CYP3A activity in human liver microsomes lead to a variable metabolism of most CYP3A substrates[26]. It was reported that the VRL metabolism varied approximately 10-fold between individuals[23]. As the structure and metabolism of vinflunine is similar to VRL, interindividual variability of the vinflunine metabolism needs to be investigated. In conclusion, the present study indicates that the vinflunine metabolism may be affected by the drugs that have an inhibitory or inductive effect on CYP3A4, because CYP3A4 plays a major role in the metabolism of vinflunine in human liver microsomes.
References
- Jordan MA, Thrower D, Wilson L. Mechanism of inhibition by Vinca alkaloids. Cancer Res 1991;51:2212-22.
- Wilson L, Panda D, Jordan MA. Modulation of microtubule dynamics by drugs: a paradigm for the actions of cellular regulators. Cell Struct Funct 1999;24:329-35.
- Fahy J, Duflos A, Ribet JP. Vinca alkaloids in superacidic media: a method for creating a new family of antitumor derivatives. J Am Chem Soc 1997;119:8576-7.
- Bunn PA., Kelly K. New chemotherapeutic agents prolong survival and improve quality of life in non-small cell lung cancer: a review of the literature and future directions. Clin Cancer Res 1998;4:1087-100.
- Crown J. Optimising treatment outcomes: a review of current management strategies in first-line chemotherapy of metastatic breast cancer. Eur J Cancer 1997;33:S15-9.
- Johnson SA, Harper P, Hortobagyi GN, Pouillart P. Vinorelbine: an overview. Cancer Treat Rev 1996;22:127-42.
- Kruczynski A, Colpaert F, Tarayre JP, Mouillard P, Fahy J, Hill BT. Preclinical in vivo antitumor activity of vinflunine, a novel fluorinated Vinca alkaloid. Cancer Chemother Pharmacol 1998;41:437-47.
- Hill BT, Fiebig HH, Waud WR, Poupon MF, Colpaert F, Kruczynski A. Superior in vivo experimental antitumour activity of vinflunine, relative to vinorelbine, in a panel of human tumour xenografts. Eur J Cancer 1999;35:512-20.
- Kruczynski A, Hill BT. Vinflunine, the latest Vinca alkaloid in clinical development. A review of its preclinical anticancer properties. Crit Rev Oncol Hematol 2001;40:159-73.
- Jordan MA, Wilson L. Kinetic analysis of tubulin exchange at microtubule ends at low vinblastine concentrations. Biochemistry 1990;29:2730-9.
- Kruczynski A, Barret JM, Etievant C, Colpaert F, Fahy J, Hill BT. Antimitotic and tubulin-interacting properties of vinflunine, a novel fluorinated Vinca alkaloid. Biochem Pharmacol 1998;55:635-48.
- Singer WD, Himes RH. Cellular uptake and tubulin binding properties of four Vinca alkaloids. Biochem Pharmacol 1992;43:545-51.
- Bennouna J, Fumoleau P, Armand JP, Raymond E, Campone M, Delgado FM, et al. Phase I and pharmacokinetic study of the new vinca alkaloid vinflunine administered as a 10-min infusion every 3 weeks in patients with advanced solid tumours. Ann Oncol 2003;14:630-7.
- Holwell SE, Hill BT, Bibby MC. Anti-vascular effects of vinflunine in the MAC 15A transplantable adenocarcinoma model. Br J Cancer 2001;84:290-5.
- Etiévant C, Barret JM, Kruczynski A, Perrin D, Hill BT. Vinflunine (20',20'-difluoro-3',4'-dihydrovinorelbine), a novel Vinca alkaloid, which participates in P-glycoprotein (Pgp)-mediated multidrug resistance in vivo and in vitro. Invest New Drugs 1998;16:3-17.
- Jacquesy JC, Jouannetaud MP. Vinflunine: a new anti-cancer fluorinated agent derived from Vinca-alkaloids. Ann Pharm Fr 2005;63:28-34.
- Zhou XJ, Placidi M, Rahmani R. Uptake and metabolism of vinca alkaloids by freshly isolated human hepatocytes in suspension. Anticancer Res 1994;14:1017-22.
- Zhou XJ, Zhou-Pan XR, Gauthier T, Placidi M, Maurel P, Rahmani R. Human liver microsomal cytochrome P450 3A enzymes mediated vindesine biotransformation: Metabolic drug interactions. Biochem Pharmacol 1993;45:853-61.
- Zhou-Pan XR, Seree E, Zhou XJ, Placidi M, Maurel P, Barra Y, et al. Involvement of human liver cytochrome P450 3A in vinblastine metabolism: Drug interactions. Cancer Res 1993;53:5121-6.
- Van Heugen JC, De Graeve J, Zorza G, Puozzo C. New sensitive liquid chromatography method coupled with tandem mass spectrometric detection for the clinical analysis of vinorelbine and its metabolites in blood, plasma, urine and faeces. J Chromatogr A 2001;926:11-20.
- Yamaguchi K, Yasuzawa T, Sakai T, Kobayashi S. Identification of novel metabolites of vinorelbine in rat. Xenobiotica 1998;28:281-91.
- Kajita J, Kuwabara T, Kobayashi H, Kobayashi S. CYP3A4 is mainly responsible for the metabolism of a new vinca alkaloid, vinorelbine, in human liver microsomes. Drug Metab Dispos 2000;28:1121-7.
- Dominique BR, Pascale G, Christian P, Francois S, Christian F, Christian R, et al. Characterization of human cytochrome P450 isoenzymes involved in the metabolism of vinorelbine. Fundam Clin Pharmacol 2005;19:545-53.
- Liu XQ, Zhao Y, Li D, Qian ZY, Wang GJ. Metabolism and metabolic inhibition of clinipine in human liver microsomes. Acta Pharmacol Sin 2003;24:263-8.
- Zhao XP, Liu XQ, Wang YS, Zhong J, Chen Y, Wang GJ. Determination of vinflunine in rat plasma by liquid chromatography-electrospray ionization mass spectrometry for a pharmacokinetic study. J Pharm Biomed Anal 2006;41:906-11.
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Inter-individual variations in human liver cytochrome P450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 1994;270:414-23.