
Triptolide suppresses IL-1β-induced chemokine and stromelysin-1 gene expression in human colonic subepithelial myofibroblasts1
Introduction
Crohn’s disease (CD) and ulcerative colitis are chronic inflammatory bowel diseases (IBD) of unknown aetiology. Up to now, few therapies have been shown to reliably cure them, especially CD[1].
Mucosa ulcer and tissue fibrosis are frequently observed in IBD, and are thought to develop as a result of abnormal wound repair following inflammatory responses. Chemokines and matrix metalloproteinases (MMP) expressed by subepithelial myofibroblasts (SEMF)[2] play important roles in these processes by regulating inflammatory responses and extracellular matrix metabolism[3]. MMP-3 (stromelysin-1) has a profound immunoregulatory impact on intestinal immune activation[4] and is proven to be a key enzyme for degradation of the lamina propria extracellular matrix[5]. Chemokines expressed by SEMF such as interleukin (IL)-8 and monocyte chemotactic protein (MCP)-1, participate in inflammation by inducing an accumulation of leukocytes to the inflammatory sites[6]. These create a plausible link between mucosal inflammation and destruction of the subepithelial matrix. Thus, inhibition of these processes represents a lucrative target for IBD therapies.
Triptolide is an extract of Tripterygium wilfordii Hook F, the traditional Chinese medicine, which has both anti-inflammatory and immunomodulatory activities. Triptolide inhibits lymphocyte proliferation, synthesis and secretion of pro-inflammatory cytokines[7,8]. Triptolide also induces apoptosis in T cells[9]. Recent studies have shown that triptolide inhibits dendritic cell-mediated chemo-attraction of neutrophils and T cells through inhibiting Stat3 phosphorylation and nuclear factor (NF)-κB activation[10]. Previous studies also show triptolide as a potent inhibitor of chemokine and MMP[11,12]. However the data of its effects on intestinal cells are lacking. Okuno et al[13] demonstrated that IL-1β induces chemokine and MMP-3 expressions in SEMF through the NF-κB signal pathway. Triptolide was demonstrated as a selected NF-κB inhibitor[7,8]. Therefore we undertook the present study to evaluate the role of triptolide in the regulation of chemokine (IL-8 and MCP-1) and MMP-3 in SEMF stimulated with IL-1β.
Materials and methods
Reagents Crystalline triptolide (PG490, molecular weight 360, purity 99%) was obtained from the Institute of Derma-tology, Chinese Academy of Medical Sciences (Nanjing, China). Triptolide was dissolved in dimethyl sulfoxide (DMSO) and stock solutions (1 mg/mL) stored at -20 °C. Triptolide was freshly diluted to the indicated concentration with culture medium before use in experiments. DMSO concentration in test conditions did not exceed 0.001% (v/v). Human recombinant IL-1β was purchased from Sigma (St Louis, MO, USA). IL-1β was reconstituted in phosphate buffered saline (PBS). Antibody for phosphorylated IκB-α was purchased from Cell Signaling (Beverly, MA, USA). Secondary goat anti-rabbit antibody were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA). All the other reagents were purchased from Sigma unless stated otherwise.
Culture and characterization of SEMF The primary cultures of SEMF were performed according to the method described by Mahida et al[14]. In brief, mucosa samples were obtained from surgical specimens (>5 cm from the tumor margin) from patients undergoing a partial colectomy for carcinomas, with their informed consent. The mucosa samples were completely denuded of epithelial cells by 3 30 min incubations at 37 °C in 1 mmol/L EDTA (Sigma, USA). The de-epithelialized mucosal samples were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 50 U/mL penicillin and 50 µg/mL streptomycin (all from GIBCO-BRL, Gaithersburg, MD, USA), and incubated at 37 °C in 5% CO2-atmosphere. The cells in suspension were removed after every 24–72 h culture period, and the denuded mucosal tissue was maintained in culture for up to 4 weeks. Established colonies of myofibro-blasts were cultured in DMEM containing 10% FBS, 50 U/mL penicillin and 50 µg/mL streptomycin. Before stimulation in all of the following experiments, the culture medium was replaced with serum-free DMEM for 24 h. The studies were performed on passages 2–6 of myofibroblasts isolated from 9 resection specimens.
Cultured SEMF were characterized with immunohistochemistry and transmission electron microscopy. Mouse monoclonal antibodies to α-smooth muscle actin and vimentin were used. The cells were grown on glass coverslips and fixed with acetone before immunoperoxidase staining with the Vectastain ABC peroxidase kit (Vecta Laboratories, Burlingame, CA, USA). After incubation with the primary antibody, biotinylated goat anti-mouse IgG was applied, followed by avidin-biotinylated horseradish peroxidase com-plex. Peroxidase activity was developed with diaminobenzidine, followed by nuclear staining using hematoxylin.
The cultured cells were also studied by transmission electron microscopy (JEM-1200 EX, Joel, Tokyo, Japan). The cells were fixed by immersion in 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4) for 2 h. Suitable areas were then selected, and 18 nm sections were cut and mounted on copper grids.
Triptolide toxicity assays on SEMF The viability affected by triptolide was evaluated using MTT assay. The cells seeded in 96-well plates (Coster, Cambridge, MA, USA) with 1×103–5×103 cells per well were incubated with various concentrations of triptolide (0.1, 1, 10, 30, 60, 100, 150, 200, and 400 ng/mL) for 24 h. The cells were then incubated with 200 µL of media containing 20 µL of MTT solution for 4 h. The media were removed, the precipitated fomazan was solubilized with DMSO and absorbance was measured at 490 nm using a microplate reader (Sunrise, Tecan, Austria). The cell survival rate (CSR)=[Optical Density (OD) value in experimental group – OD value in blank control group]/(OD value in control group – OD value in blank control group)×100%. Values represent the mean±SD from 3 separate experiments.
The concentrations of triptolide chosen to be used in the present study induced less than 10% reduction of the cell survival rate. The MTT results indicated that incubation with triptolide (0.1, 1, 10, 30, and 60 ng/mL) for 24 h was not detrimental to cell viability (CSR >90%). Incubation with 100 ng/mL triptolide decreased cell viability (CSR=84.0%±5.1%), and greater toxicity was observed at doses above 100 ng/mL triptolide (CSR=74.5%±3.6%, 58.6%±4.5%, and 41.1%±5.9%, respectively). Therefore, the doses of triptolide used in all of the following experiments were 0.1–60 ng/mL to exclude toxicity.
Assay of chemokine and MMP-3 secretion The release of IL-8, MCP-1, and MMP-3 was determined by ELISA. The culture medium at the designated time was collected and centrifuged at 1000×g for 5 min, and the resultant supernatant was frozen at -80 °C for subsequent measurement of MMP-3, IL-8 (both from Biosource, Camarillo, CA, USA) and MCP-1 (Bender, Vienna, Austria) concentrations. The concentrations were normalized by expression as nanograms per 1×105 cells.
RNA extraction and quantitative RT-PCR analysis The mRNA expression of IL-8, MCP-1, and MMP-3 was determined by RT-PCR. At the designated time, the cells were harvested with 0.25% trypsin and 0.02% EDTA and the total RNA was isolated using RNeasy reagents (Qiagen, Chats-worth, CA, USA) according to the manufacturer’s protocol and quantitated by spectrophotometry. For synthesization of cDNA, 1 µg of total RNA was treated with reverse transcriptase (Promega, Madison, WI, USA) and oligo (dT) were used for reverse transcription. Reactions were performed using the Reverse Transcription System (Promega, USA) under the following conditions: 42 °C for 15 min, 95 °C for 5 min and 4 °C for 5 min. The samples were stored at -20 °C until use.
Real-time quantitative Taqman PCR analysis was performed using an ABI PRISM 7700 (Perkin-Elmer, Foster City, CA, USA). Specific primers and dual-labeled fluorescent probes were designed using the Primer Express primer design program v1.01 (Perkin-Elmer, USA). The constitutively expressed β-actin was used as an internal control. Probes were labeled with the fluorescent reporter dye 5-carboxyfluo-rescein (FAM) at the 5' end and the quencher N,N,N,N'-tetramethyl-6-carboxyrhodamine (TAMRA) or minor groove binder (MGB) at the 3' end. The primer and probe sequences were as follows: MMP-3 forward primer (FP) 5'-AAG ACT TTC CAG GGA TTG ACT CAA-3', reverse primer (RP) 5'-TGT GTC ACT TTC TTT GCA TTT GG-3' and probe 5'-(FAM)-AAA GAA CCC AAA TTC TTC AAA-(MGB)-3'; β-actin FP 5'-CAT TGC CGA CAG GAT GCA-3', RP 5'-CAT CTG CTG GAA GGT GGA CAG -3' and probe 5'-(FAM)-AGC AAT GAT CTT GAT CTT CA-(MGB)-3'. IL-8 (CXCL8) FP 5'-GCT CTC TTG GCA GCC TTC CT-3', RP 5'-TGG AAA GGT TTG GAG TAT GTC TTT ATG-3' and probe 5'-(FAM)-ACT GCA CCT TCA CAC AGA GCT GCA GAA-(TAMRA)-3'; β-actin FP 5'-CCT GGC ACC CAG CAC AAT-3', RP 5'-GCT GAT CCA CAT CTG CTG GAA-3' and probe 5'-(FAM)-ATC AAG ATC ATT GCT CCT CCT GAG CGC-(TAMRA)-3'. MCP-1 (CCL2) FP 5'-CAT AGC AGC CAC CTT CAT TCC-3', RP 5'-TCT GCA CTG AGA TCT TCC TAT TGG T-3' and probe 5'-(FAM)-CTC GCT CAG CCA GAT GCA ATC AAT G-(TAMRA)-3'; β-actin primer and probe were as same as in the PCR analysis for IL-8.
Samples were amplified in a final volume of 25 µL. Primers were used at a concentration of 900 nmol/L, and probes at 250 nmol/L. β-actin was amplified in separate reactions. The cycling conditions were as follows: 50 °C for 2 min, 95 °C for 10 min, 45 cycles of 95 °C for 30 s and 60 °C for 30 s. Data were normalized to β-actin gene expression and shown as fold increases in expression.
Cytosolic and nuclear extracts Cytosolic and nuclear extracts were prepared as described by Finto et al[15]. The cells were washed in cold PBS and resuspended in 500 µL of cold buffer [10 mmol/L hydroxyethyl piperazine ethane-sulfonic acid (HEPES), 2 mmol/L MgCl2, 0.1 mmol/L EDTA, 10 mmol/L KCl, 1 mmol/L dithiothreitol, 0.5 mmol/L phenyl-methylsulfonyl fluoride, 1 µg/mL leupepstatin, 1 µg/mL pepstatin and 1 µg/mL leucine thiol, 0.1% IgePal CA 630, pH 7.9] and left on ice for 30 min. The samples were then mixed and centrifuged at 4000×g for 30 min. The supernatant (cytosolic extract) was kept at -70 °C. The pellet was resuspended in 150 µL of cold saline buffer [20 mmol/L HEPES, 50 mmol/L KCl, 0.1 mmol/L EDTA, 1.5 mmol/L MgCl2, 300 mmol/L NaCl, 25% (w/v) glycerol, 1 mmol/L dithiothreitol, 0.5 mmol/L phenylmethylsulfonyl fluoride, 1 µg/mL leupepstatin, 1 µg/mL pepstatin and 1 µg/mL leucine thiol, 0.2% IgePal CA 630, pH 7.9] and left on ice for 1 h. After centrifuging at 12 000×g for 15 min at 4 °C, the supernatant (nuclear extract) containing the nuclear proteins was collected and stored at -70 °C. Protein content was measured by Modified lowry protein assay kit (Pierce, Rockford, IL, USA).
Western immunoblotting The cytosolic amount of p-IκB-α was determined by Western blotting. Equal amounts of protein in the sample were first resolved by SDS-PAGE, then transferred electrophoretically to the nitrocellulose membrane and subsequently incubated with the primary antibody. For detection, the nitrocellulose filter was incubated with a horseradish peroxidase coupled secondary antibody, followed by an enhanced chemiluminescence substrate reaction using ECL Western blotting detection reagents (Amersham Pharmacia Biotech, USA).
Electrophoretic mobility shift assay (EMSA) DNA binding activity of NF-κB was characterized by EMSA performed using a commercial kit (Gel Shift Assay System, Promega, USA). Double stranded consensus oligonucleotide (5'-AGT TGA GGG GAC TTT CCC AGG C-3') was 5'-end-labeled with T4 polynucleotide kinase and [γ-32P] ATP (Promega, USA). Nuclear protein (10 µg) was pre-incubated in a total volume of 9 µL in a binding buffer, consisting of 10 mmol/L Tris-HCl (pH 7.5), 4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 0.5 mmol/L NaCl, and 0.05 mg/mL poly(di-dc) for 10 min at room temperature. After the addition of the 32P-labled oligonucleotide probe, the incubation was continued for 20 min at room temperature. Reaction was stopped by adding 1 µL of gel-loading buffer and the mixture was subjected to nondenaturing 4% polyacrylamide gel electrophoresis in 0.5×TBE buffer (Tris-borate-EDTA). After electrophoresis was conducted at 390 V for 1 h, the gel was vacuum-dried and exposed to X-ray film (Fuji Hyperfilm, Tokyo, Japan) at -70 °C with an intensifying screed.
Statistical analysis Data are expressed as mean±SD. Statistical significance between 2 groups was tested using the unpaired Student’s t test. Statistical significance between more than 2 groups was tested using one-way ANOVA followed by the Student-Newman-Keuls test. Statistical significance was set at P<0.05.
Results
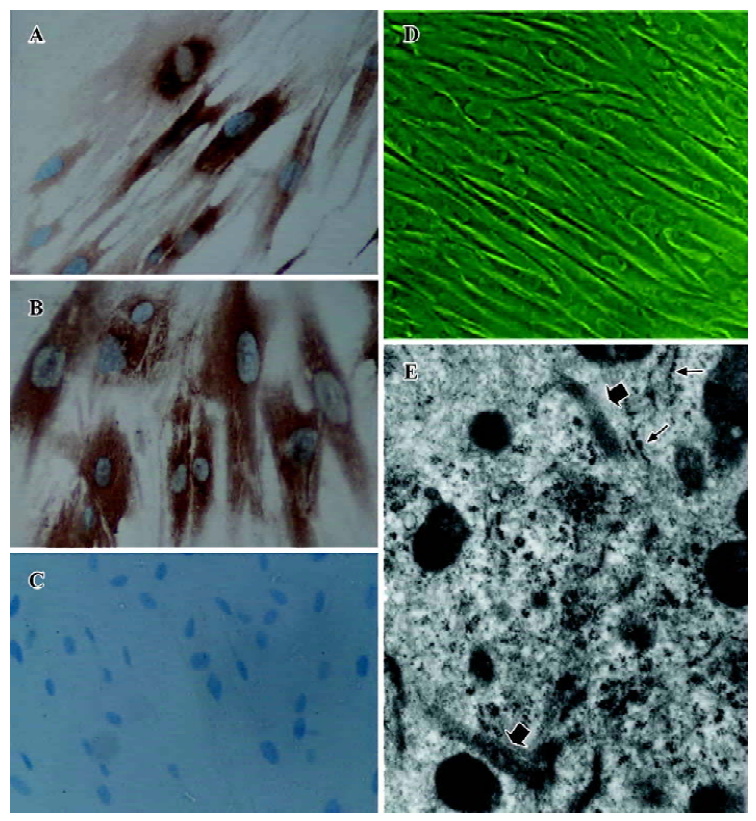
Culture and characterization of SEMF Colonic subepithelial myofibroblast cultures were successfully established from 9 resection specimens. The studies were performed on passages 2–6. The cultures obtained were pure myofibro-blast monolayers with no evidence of any contaminating lamina propria or epithelial cells (Figure 1). Immunohistochemical studies of SEMF showed that the cells expressed the immunoreactivity for α-SMA and vimentin (Figure 1A, 1B). Filamentous immunostaining was clearly observed. Ultrastructu
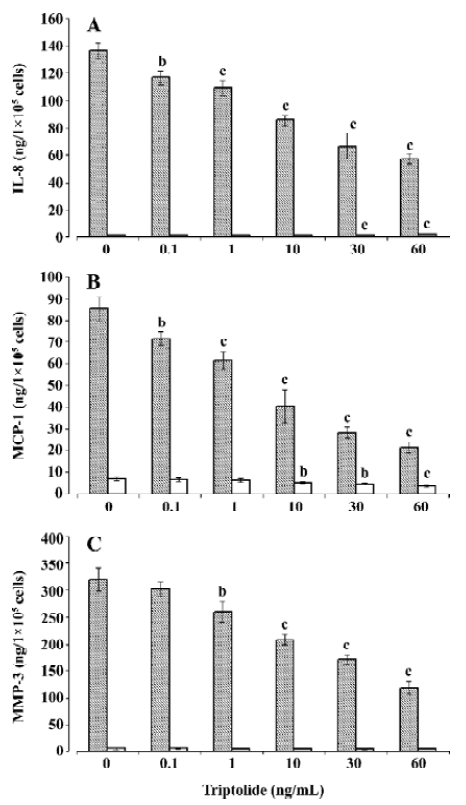
Effects of triptolide on chemokine and MMP-3 release To examine the modulating role of triptolide on chemokine and MMP-3, the cells seeded in 6-well plates with 1×105–1×106 cells per well were incubated with various concentrations of triptolide (0.1–60 ng/mL) or equivalent amount of its vehicle (DMSO; control) for 1 h and then in the additional IL-1β (10 ng/mL) or not (triptolide-alone-treatment) for 24 h. As shown in Figure 2A, 2B, triptolide induced an inhibitory effect on basal IL-8 (2.18±0.02 ng/1×105 cells per 24 h) and MCP-1 release (6.96±0.87 ng/1×105 cells per 24 h) above a concentration of 30 and 10 ng/mL, respectively, but triptolide at any concentration did not significantly modify the basal release of MMP-3 (7.25±2.04 ng/1×105 cells per 24 h; Figure 2C). We next investigated the effects of triptolide on chemokine and MMP-3 release stimulated by IL-1β. Incubation in the presence of IL-1β (10 ng/mL) resulted in approximately 60-, 15-, and 45-fold increases in the release of IL-8, MCP-1, and MMP-3, respectively (Figure 2). Triptolide significantly inhibited these effects of IL-1β in a dose-dependent manner. At a concentration of 60 ng/mL, triptolide inhibited the IL-1β-induced release of IL-8, MCP-1, and MMP-3 by approximately 60%, 75%, and 60%, respectively.
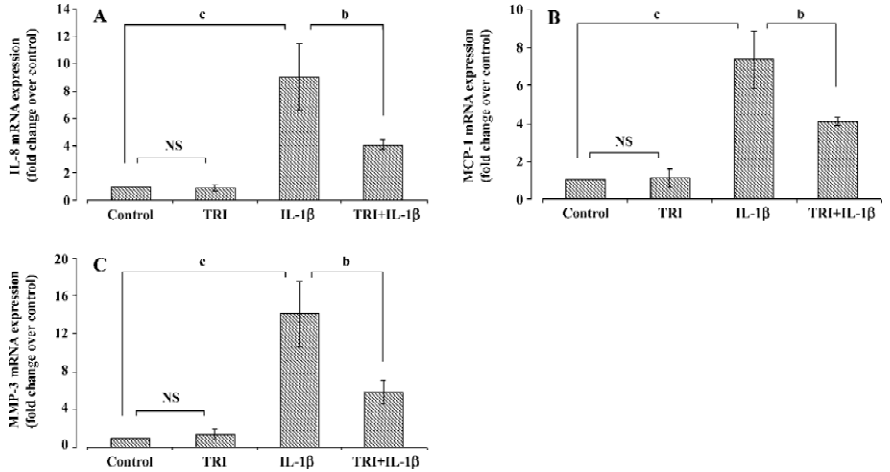
Effects of triptolide on chemokine and MMP-3 mRNA expression The cells were incubated with triptolide (60 ng/mL) or an equivalent amount of DMSO for 1 h and then in the additional IL-1β (10 ng/mL) or not for 24 h. RT-PCR analysis demonstrated that IL-1β (10 ng/mL) induced 9-, 7-, and 14-fold increases in the amounts of IL-8, MCP-1, and MMP-3 mRNA, respectively. Triptolide (60 ng/mL) did not modify the basal chemokine or MMP-3 mRNA expression in SEMF (P>0.05), but it diminished the upregulation effect of IL-1β on IL-8 (P<0.05), MCP-1 (P<0.05), and MMP-3 (P<0.01) mRNA expression (Figure 3).
Effects of triptolide on NF-κB DNA binding activity Previous studies have demonstrated that the IL-1β-induced expression of IL-8, MCP-1, and MMP in SEMF is regulated by the activation of NF-κB[13]. Therefore, we investigated the effect of triptolide on IL-1β-induced NF-κB DNA binding activity. The cells were incubated with triptolide (60 ng/mL) or an equivalent amount of DMSO for 1 h and then in the additional IL-1β (10 ng/mL) or not for 1 h. As shown in Figure 4A, the DNA binding capacity of NF-κB in the resting SEMF was very low and was unregulated by triptolide. Stimulation with IL-1β (10 ng/mL) induced approximately 3-fold increase in the DNA binding capacity of NF-κB, and triptolide (60 ng/mL) inhibited this effect by more than 70% (Figure 4A). Thus, triptolide may repress chemokine and MMP-3 expression partly by interfering NF-κB activity.
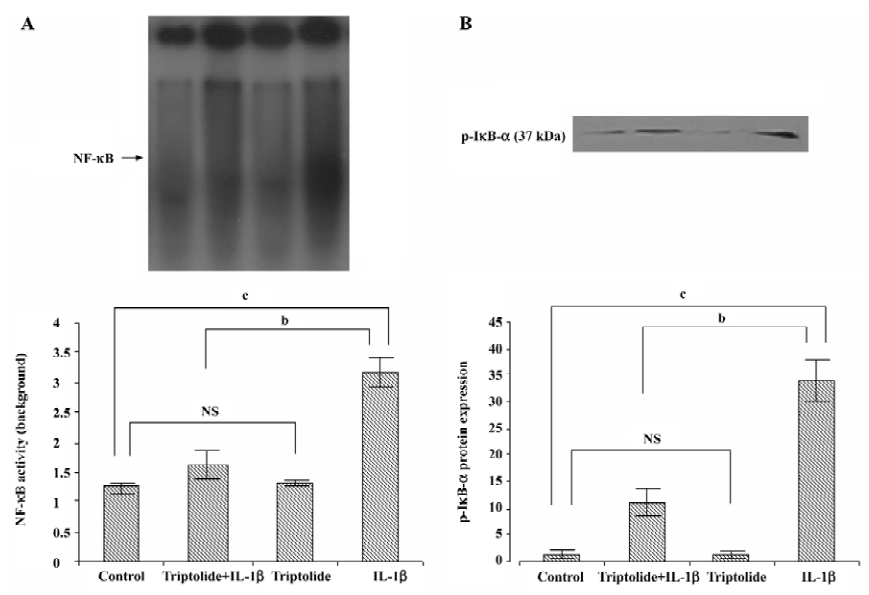
Effects of triptolide on IL-1β-induced phosphorylation of IκB-α To further reveal the effect of triptolide on the signal pathway of NF-κB in IL-1β-stimulated SEMF, we investigated the p-IκB-α levels in the cytosolic extract. The cells were incubated with triptolide (60 ng/mL) or an equivalent amount of DMSO for 1 h and then in the additional IL-1β (10 ng/mL) or not for 1 h. As shown in Figure 4B, the levels of p-IκB-α were increased upon IL-1β stimulation; the levels were significantly suppressed with triptolide pretreatment (P<0.05). Measurement of band densities indicated that triptolide pretreatment inhibited IL-1β-induced increase of p-IκB-α by 60%. Therefore, triptolide might inhibit DNA binding activity of NF-κB by preventing the phosphorylation of IκB-α.
Discussion
We report here for the first time that triptolide inhibits IL-1β-induced chemokine and MMP expression by inhibiting NF-κB activity in human colonic subepithelial myofibroblasts. This may be a potential mechanism by which triptolide protects extracellular matrix under inflammatory conditions and becomes a potential therapeutic agent for treatment of IBD.
During inflammation in the colon, several chemokines may be secreted locally by colonic subepithelial myofibro-blasts in response to IL-1β and TNF-α, and thus might induce the subsequent infiltration and activation of leukocytes into the sites of inflammation and result in the continuous activation and amplification of the cytokine cascade. In inflammatory and ulcerated lesions of IBD, MMP-3 mRNA, which encodes a key enzyme in matrix degradation, is highly increased, paralleled by an increase in MMP-3 protein levels[16].
The mechanisms of action of triptolide are not known. Our work has characterized chemokine and MMP as novel targets of its therapeutic actions which is in agreement with the previous findings[12,13]. The fact that both mRNA and protein are inhibited in a similar fashion suggests that the inhibition of chemokine and MMP is at the transcriptional or pretranscriptional levels. NF-κB plays an important role in intracellular signaling induced by cytokines and is therefore a potential target for new therapeutic approaches to inflammatory diseases. NF-κB is maintained inactive in the cytoplasm through its association with an inhibitory subunit, Iκ-B (α or β). In response to an extracellular signal, such as IL-1 or TNF-α, Iκ-B is phosphorylated, polyubiquitinated and targeted to the proteasome, where it is degraded[17]. This degradation unmasks the nuclear localization sequence of NF-κB, which is rapidly translocated to the nucleus, where it binds to specific nucleotide sequences. This binding recruits the RNA polymerase complex and leads to the specific transcription of several genes involved in the pro-inflammatory response including chemokines, transcription factor-associated proteins, cell adhesion molecules, cytokines and immunoreceptors[17]. MMP promoters have AP-1 and c-ets-1 transcription factor binding sites[18]. Further NF-κB may also be indirectly implicated in MMP gene regulation[19,20]; the link between NF-κB and chemokine and MMP has been experimentally demonstrated[13]. In the current study, triptolide inhibited the activation of NF-κB induced by IL-1β in SEMF, suggesting this effect may contribute to its inhibition of IL-1β-induced chemokine and MMP-3 expression.
Triptolide has several characteristics of particular interest for IBD. It clearly has anti-inflammatory and immunosuppressive effects. It inhibits several pro-inflammatory cytokines and adhesion molecules, which are all important mediators of IBD. Triptolide has been shown to be safe and clinically beneficial in rheumatoid arthritis[21]. It has also been shown to be effective in the treatment of several autoimmune diseases such as lung fibrosis[22] and uveoretinitis[23] in animal models. Yan et al[24] showed triptolide could inhibit IFN-γ-induced activation of fibroblasts derived from patients with Graves’ ophthalmopathy. Steroids have been administered widely for their anti-inflammatory activity in IBD, but they are not free of adverse effects. Such adverse reactions may be avoided if triptolide proves effective for the treatment of IBD, particularly for CD. The present study indicates that triptolide could be a potential therapeutic agent for IBD by its extracellular matrix protective and anti-inflammatory properties. Whether triptolide is a viable alternative to steroids for the treatment of CD and is devoid of adverse effects remain to be clarified.
In summary, triptolide inhibits IL-1β-induced chemokine and MMP-3 expression by colonic subepithelial myofibro-blast at both mRNA and protein levels. Inhibition of NF-κB activity is accompanied with and may contribute to the effects.
References
- Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut 2004;53 Suppl 5:V1-16.
- Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB II. Intestinal subepithelial myofibroblasts. Am J Physiol 1999;277:C183-201.
- McKaig BC, McWilliams D, Watson SA, Mahida YR. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol 2003;162:1355-60.
- Kruidenier L, Macdonald TT, Collins JE, Pender SLF, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology 2006;130:127-36.
- Salmela MT, MacDonald TT, Black D, Irvine B, Zhuma T, Saarialho-Kere U, et al. Upregulation of matrix metallopro-teinases in a model of T cell mediated tissue injury in the gut: analysis by gene array and in situ hybridization. Gut 2002;51:540-7.
- Banks C, Bateman A, Payne R, Johnson P, Sheron N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J Pathol 2003;199:28-35.
- Qiu D, Zhao G, Aoki Y, Shi L, Uyei A, Nazarian S, . Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-kappaB transcriptional activation. J Biol Chem 1999; 274: 13 443–50.
- Krakauer T, Chen X, Howard OM, Young HA. Triptolide attenuates endotoxin- and staphylococcal exotoxin-induced T-cell proliferation and production of cytokines and chemokines. Immuno-pharmacol Immunotoxicol 2005;27:53-66.
- Yang Y, Liu Z, Tolosa E, Yang J, Li L. Triptolide induces apoptotic death of T lymphocyte. Immunopharmacology 1998;40:139-49.
- Liu Q, Chen T, Chen G, Li N, Wang J, Ma P, et al. Immunosuppressant triptolide inhibits dendritic cell-mediated chemoattrac-tion of neutrophils and T cells through inhibiting Stat3 phosphorylation and NF-kappaB activation. Biochem Biophys Res Commun 2006;345:1122-30.
- Liacini A, Sylvester J, Zafarullah M. Triptolide suppresses proinflammatory cytokine-induced matrix metalloproteinase and aggrecanase-1 gene expression in chondrocytes. Biochem Biophys Res Commun 2005;327:320-7.
- Lu Y, Fukuda K, Nakamura Y, Kimura K, Kumagai N, Nishida T. Inhibitory effect of triptolide on chemokine expression induced by proinflammatory cytokines in human corneal fibroblasts. Invest Ophthalmol Vis Sci 2005;46:2346-52.
- Okuno T, Andoh A, Bamba S, Araki Y, Fujiyama Y, Fujimiya M, et al. Interleukin-1β and tumor necrosis factor-α induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol 2002;37:317-24.
- Mahida YR, Beltinger J, Marh S, Goke M, Gray T, Podolsky DK, et al. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase-1 and -2. Am J Physiol 1997;273:G1341-8.
- Finco TS, Beg AA, Baldwin AS Jr. Inducible phosphorylation of IκBα is not sufficient for its dissociation from NF-κB and is inhibited by protease inhibitors. Proc Natl Acad Sci USA 1994;91:11884-8.
- Baugh MD, Perry MJ, Hollander AP, Davies DR, Cross SS, Lobo AJ, et al. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology 1999;117:814-22.
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol 1994;10:405-55.
- Pendas AM, Balbin M, Llano E, Jimenez MG, Lopez-Otin C. Structural analysis and promoter characterization of the human collagenase-3 gene (MMP13). Genomics 1997;40:222-33.
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metallo-proteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum 2000;43:801-11.
- Pattoli MA, MacMaster JF, Gregor KR, Burke JR. Collagen and aggrecan degradation is blocked in interleukin-1-treated cartilage explants by an inhibitor of IkappaB kinase through suppression of metalloproteinase expression. J Pharmacol Exp Ther 2005;315:382-8.
- Tao X, Younger J, Fan FZ, Wang B, Lipsky PE. Benefit of an extract of Tripterygium wilfordii Hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum 2002;46:1735-43.
- Krishna G, Liu K, Shigemitsu H, Gao M, Raffin TA, Rosen GD. PG490-88, a derivative of triptolide, blocks bleomycin-induced lung fibrosis. Am J Pathol 2001;158:997-1004.
- Wu Y, Wang Y, Zhong C, Li Y, Li X, Sun B. The suppressive effect of triptolide on experimental autoimmune uveoretinitis by down-regulating Th1-type response. Int Immunopharmacol 2003;3:1457-65.
- Yan SX, Wang Y. Inhibitory effects of triptolide on interferon-gamma-induced human leucocyte antigen-DR, intercellular adhesion molecule-1, CD40 expression on retro-ocular fibroblasts derived from patients with Graves’ ophthalmopathy. Clin Exp Ophthalmol 2006;34:265-71.