
Apoptosis initiated by carbon tetrachloride in mitochondria of rat primary cultured hepatocytes
Introduction
Apoptosis is a genetically encoded form of cell suicide central to the development and homeostasis of multicellular organisms[1–3]. Once researchers assumed that the activation of endonucleases and specific proteases (such as caspases) reflect the key mechanism of apoptosis[4,5]. However, a number of studies disclose that mitochondria play a key role in apoptosis[1,4,6]. The mitochondrial pathway is partly dependent on the release of cytochrome c. After release from mitochondria to the cytosol, cytochrome c binds to apoptosis-activating factor-1 (Apaf-1), ATP (or dATP), and possibly a cytosolic protein (Apaf-3), and activates caspase 9, which in turn stimulates caspase 3 activity. Caspase 3 cleaves and activates DNA fragmentation factor (DFF), resulting in DNA degradation[2,4,7].
Bcl-2 proteins act on mitochondria to regulate apoptosis. The Bcl-2 family consists of both cell death promoters and preventers, including the anti-apoptotic proteins Bcl-2, Bcl-XL, Mcl-1, A1/Bfl-1 and Bcl-W, and the pro-apoptotic members Bax, Bcl-XS, Bak, Bad, Bik, Bim, Bid, Hrk and Bok[8]. Bcl-2 and Bcl-XL prevent cytochrome c from entering the cytosol, either by blocking release or binding to the cytochrome in a direct or indirect fashion, and consequently inhibiting activation of the downstream caspase cascade[4].
Reactive oxygen species (ROS), which induce the onset of the mitochondrial permeability transition (MPT), play an important role in mitochondrial apoptosis. Activation of MPT is a major controlling mechanism in some apoptotic systems, and also contributes to the release of cytochrome c and other apoptogenic proteins[4,9]. However, the mechanisms of ROS generation and its relationship with the well-documented caspase activation remain to be elucidated. In the present investigation, the levels of reduced glutathione (GSH) and malondialdehyde (MDA) are measured as reliable markers of oxidative stress.
Carbon tetrachloride (CCl4) is a typical poison that induces severe oxidative stress followed by activation of caspase 3 in rat liver[10]. Moreover, GSH depletion and MDA induction by CCl4 are evident in rat primary hepatocytes and HepG2, a human hepatoma cell line[11]. However, it is currently unclear whether both oxidative stress and apoptosis occur in rat primary hepatocytes induced by CCl4.
Mitochondrial damage by CCl4 is involved in the apoptotic process in vivo, during which caspase 3 is activated[10]. A histological study suggests that apoptosis is additionally induced by CCl4 in liver[12]. However, the pathway by which CCl4 mediates apoptosis in rat primary hepatocytes is currently unknown.
In the present report, CCl4 mediation of the expression and activation of different proteins involved in apoptotic cell death is investigated. Moreover, the relationship between peroxidative damage and apoptosis induced by CCl4 in rat primary hepatocytes is explored.
Materials and methods
Materials and culture of hepatocytes CCl4 and Me2SO were purchased from Shanghai Chemical Reagent Company (Shanghai, China). CCl4 was dissolved in 10% Me2SO HBS solution (in mmol/L: HEPES 33, NaCl 160.8 mmol/L, KCl 3.15 mmol/L, Na2HPO4·12H2O 0.7 mmol/L) and diluted to final concentration of 0.1 mmol/L, 0.3 mmol/L, 1 mmol/L, 3 mmol/L, and 9 mmol/L, respectively.
Hepatocytes were isolated from Sprague-Dawley male rats (180 g–220 g) by 2-step collagenase perfusion, as described previously[13–15], with some modifications. Collagenase IV was purchased from Sigma Chemical Company (St Louis, USA). Hepatocytes were seeded on collagen-coated plastic dishes at a density of 3×105 viable cells/mL, and cultured in Ham’s F-12/Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) (1:1) medium supplemented with 15% fetal bovine serum (PAA Laboratories, Linz, Austria), 0.1 g/L penicillin (Shanghai Fourth Pharmaceuticals, Shanghai, China), 0.07 g/L streptomycin (Shanghai Fourth Pharmaceuticals, Shanghai, China) and 0.2% bovine serum albumin (Roche, NJ, USA). After 3 h, the medium was altered. Cells were incubated in medium containing different concentrations of CCl4 solution.
Cell viability and lactate dehydrogenase leakage assay Rat primary hepatocytes were treated with CCl4 for 20 h, and the general viability of cultured cells was assayed at 450 nm with the Cell Counting Kit-8 (Dojindo Laboratories, Tokyo, Japan).
Hepatocytes were seeded on collagen-coated 96-well microtiter plates at a density of 3×104 cells/well. After treatment with increasing concentrations of CCl4 for 20 h, lactate dehydrognase (LDH) released into the culture supernatants was measured in a 3-min coupled enzymatic assay that results in the conversion of a tetrazolium salt (NBT) into a formazan product[16]. The amount of color formed is proportional to the number of lysed cells. Visible wavelength absorbance data at 490 nm were collected using a standard 96-well plate reader (SOFTmax® PRO, Molecular Devices, Sunnyvale, CA, USA).
General morphology of cultured cells Hepatocytes were cultured with CCl4 for 20 h, and cell morphology was examined using inverted microscopy.
Intracellular glutathione content assay Hepatocytes were treated separately with CCl4 for 4 h, 8 h, 12 h, 16 h, or 24 h. Cells in monolayers were gently washed 3 times with phosphate-buffered saline (PBS) (pH 7.4), scraped in ice-cold PBS, and sonicated intermittently 3 times. Cell homogenates were added to solution containing 10% trichloroacetic acid and 20 mmol/L EDTA-Na2. The mixture was centrifuged for 5 min at 3000×g. The suspension was mixed with buffer(0.4 mmol/L Tris-HCl, 20 mmol/L EDTA-Na2) and 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB), and measured at a wavelength of 412 nm within 5 min. Reduced pure GSH was used to obtain a standard curve.
Lipid peroxidation assay for malondialdehyde Malondi-aldehyde levels were measured spectrophotometrically using a thiobarbituric acid (TBA) fluorescence assay. Cell homogenates were mixed with solution (20 mmol/L TBA/glacial acetic acid, 1:1), and heated for 60 min at 100 oC in a water bath. After cooling down, the mixture was extracted in methanol and centrifuged for 10 min at 5000×g. The suspension was measured in a fluorimeter with excitation at 515 nm and emission at 550 nm.
Western blot analysis Hepatocytes were scraped in ice-cold PBS obtained by centrifugation at 300×g for 5 min. Cells were gently lysed for 30 s in 75 µL ice-cold buffer containing 250 mmol/L sucrose, 1 mmol/L edetic acid, 0.05% digitonin, 25 mmol/L Tris, pH 6.8, 1 mmol/L dithiothreitol, and protease inhibitor (10-3 g/L leupeptin and aprotinin, and 0.1 mmol/L phenylmethylsulfonyl fluoride). Lysates were then homogenized through a 26-gauge needle for 30 passages and centrifuged twice at 800×g for 20 min at 4 oC. The pooled supernatant was centrifuged at 10 000×g for 10 min at 4 oC to collect the mitochondria fractions. This supernatant was then transferred to fresh tubes and centrifuged at 16 000×g for 20 min at 4 oC to remove any residual mitochondria. The supernatant was stored as the cytosolic fraction of hepatocytes. Cytosolic and mitochondria fractions were subjected to 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The gel was transferred to a PVDF membrane. Monoclonal rat anti-cytochrome c antibody was purchased from Neomarkers (Lab Vision & NEOMARKERS, UK) and polyclonal rat anti-bcl-XL antibody was from Cell Signaling Technology (Beverly, MA, USA). Following incubation with horseradish peroxidase-conjugated secondary antibody (Rockland, Gilbertsville, PA, USA), the blot was developed using the enhancing chemiluminescence detection system.
Caspase 3 activity assay Following incubation of hepatocytes in the absence of CCl4 for the indicated times, caspase 3 activation was measured with the Caspase 3 assay kit (BD Biosciences Pharmingen, San Diego, CA, USA). In brief, culture cells were washed with ice-cold PBS (pH 7.4), and lysed in buffer provided by the kit for 30 min on ice. Reaction mixtures containing Ac-DEVD-AMC and cell lysates in buffer were incubated for 1 h at 37 oC. AMC liberated from Ac-DEVD-AMC was measured in a fluorimeter with excitation at 380 nm and emission at 460 nm.
DNA gel electrophoresis assay Cultured cells were washed 3 times with ice-cold PBS, scraped in the same PBS, and collected by centrifugation for 5 min at 600×g. Cells were resuspended in 10 mmol/L EDTA and 50 mmol/L Tris-HCl (pH 8.0) containing 0.5% sodium lauryl sarcosinate and 0.5 g/L proteinase K, and incubated for 60 min at 50 oC. Next, 10 mmol/L EDTA containing 0.25% bromophenol blue and 40% sucrose was mixed with each DNA extract. Individual extracts were loaded into the wells of a 2% agarose gel containing 3×10-3 g/L ethidium bromide. Electrophoresis was carried out in 40 mmol/L Tris-HCl containing 40 mmol/L acetic acid and 1 mmol/L EDTA.
Statistical analysis Data were entered into a database and analyzed using SPSS software. Group mean values and standard deviations were calculated. After homogenetic analysis, homogeneous data were analyzed with one-way analysis of variance and a post hoc test of least significant difference. Heterogeneous data were analyzed using the t-test. P<0.05 was considered statistically significant.
Results
Cytotoxicity induction by CCl4 in rat primary hepatocytes Cells were exposed to 0.1–9 mmol/L CCl4 for 20 h, and viability was measured. Cell viability decreased in a dose-dependent manner (Table 1).
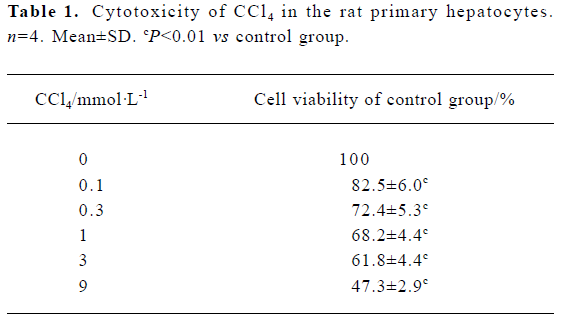
Full table
Intracellular LDH release as a result of plasma membrane breakdown and alteration of permeability was evaluated. A dose-dependent increase in LDH release in rat primary hepatocytes was observed in the presence of CCl4 (Table 2).
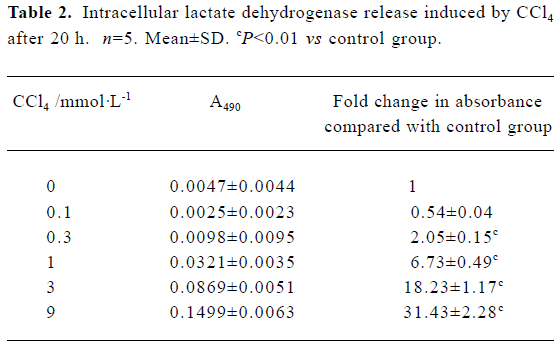
Full table
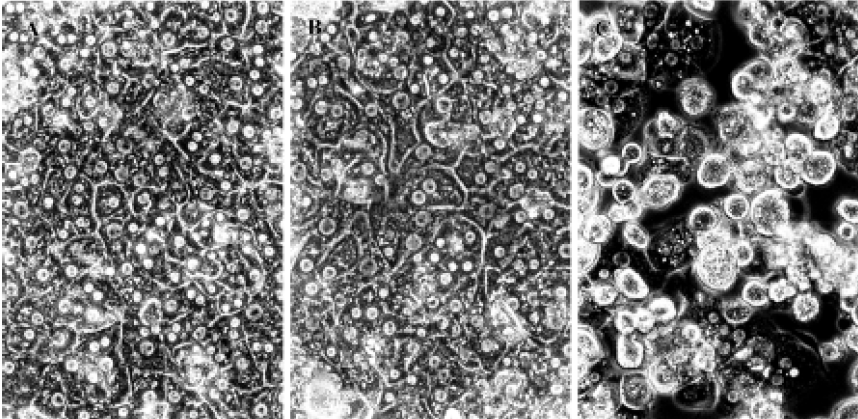
Hepatocyte morphology Cultured cell morphology examination revealed that 9 mmol/L CCl4 significantly inhibited the confluence of cultured hepatocytes (Figure 1).
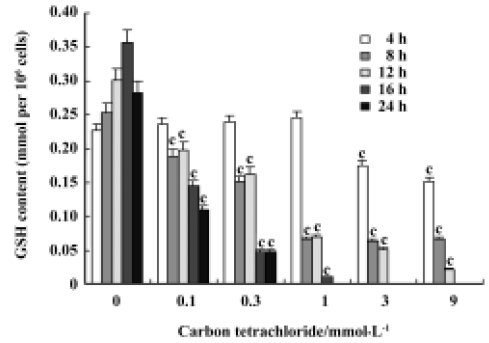
Glutathione depletion CCl4 induced a dose- and time-dependent depletion of GSH in rat primary hepatocytes. The GSH level was significantly reduced after cell incubation with 1 mmol/L CCl4 for 8 h, 12 h, 16 h, and 24 h, but not at 4 h. The exhaustion of GSH was observed initially with 3 mmol/L CCl4 after a 16-h incubation (Figure 2).
Malondialdehyde formation We observed dose- and time-dependent induction of MDA formation in the presence of CCl4 in rat primary hepatocytes. CCl4 induced significant amounts of MDA at concentrations of 3 mmol/L and 9 mmol/L after a 12-h incubation and at 0.3–9 mmol/L after 20 h. There was no evident MDA induction after 4 h of CCl4 treatment at all the doses tested (Table 3).
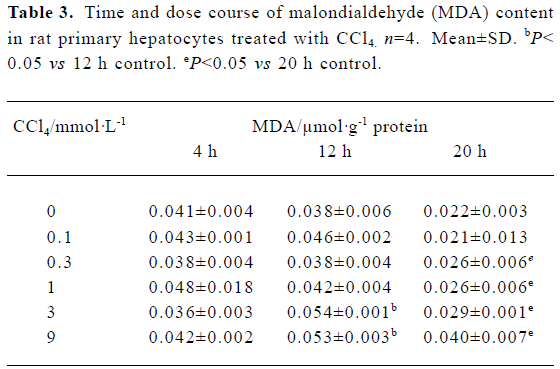
Full table
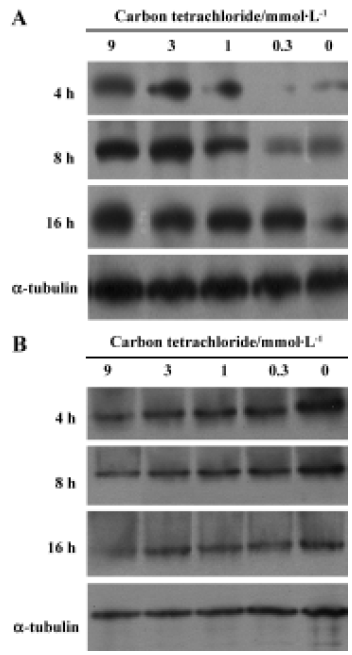
Cytochrome c release Cytochrome c was detected in the cytosolic fractions of hepatocytes. Hepatocytes were treated with 0.3, 1, 3 and 9 mmol/L CCl4 up to 16 h. CCl4 induced time-dependent cytochrome c release in rat primary hepatocytes. Evident dose-dependent release of cytochrome c was observed at 4 h and 8 h (Figure 3A). In contrast, a dose- and time-dependent decrease in mitochondria cytochrome c was observed (Figure 3B).
Caspase 3 activation by carbon tetrachloride Caspase 3 activity was examined in cultured hepatocytes treated with increasing concentrations of CCl4 for 8 h. A dose-dependent increase in caspase 3 activity was observed in the presence of 0.3–3 mmol/L CCl4, which decreased slightly at 9 mmol/L CCl4 (Table 4).
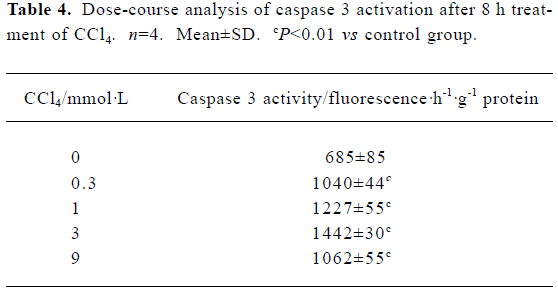
Full table
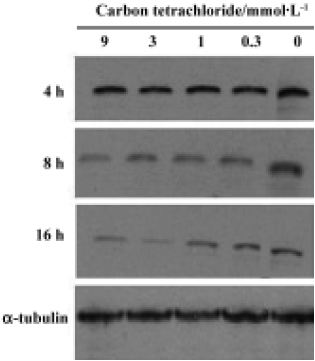
Bcl-XL analysis Bcl-XL protein levels were analyzed in cultured hepatocytes treated with different concentrations of CCl4. Hepatocytes were exposed to CCl4 for 4 h, 8 h and 16 h (Figure 4). No significant dose-dependent change was observed, but a time-dependent decrease in Bcl-XL expression was observed.
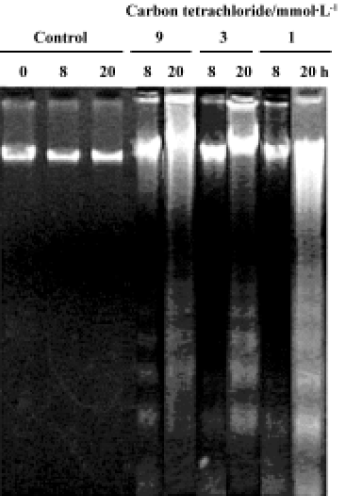
DNA fragmentation To confirm whether CCl4 induces DNA fragmentation in rat primary hepatocytes, DNA was extracted from treated cells. No ladder was observed in control cells at 0 h, 8 h, or 20 h of CCl4 treatment. Apoptosis was initially observed in the presence of 9 mmol/L CCl4 after 8 h treatment, followed by 3 mmol/L and 1 mmol/L CCl4 after 20 h treatment (Figure 5).
Discussion
Carbon tetrachloride is a typical hepatotoxin used in liver injury research. Early studies showed that the damage induced by CCl4 in liver is partly involved in the apoptosis pathway in vivo. At least 2 different apoptosis pathways – the mitochondrial pathway and the death-receptor pathway – lead to caspase activation[17]. Although past reports have disclosed caspase 3 activation and other histopathological changes in CCl4-induced apoptotic hepatocytes[10,12], little is known about the precise molecular mechanisms of apoptosis induction. In this study, we investigated the molecular mechanism of CCl4-induced apoptosis in rat primary hepatocytes. Release of cytochrome c first occurred at 4 h and then up to 16 h (Figure 3A). On the other hand, cytochrome c remaining in the mitochondria was found to decrease in a dose- and time-dependent manner (Figure 3B). In addition, it was found that caspase 3 was significantly activated after 8 h at all doses (Table 4). As a result of caspases cascade activation, DNA fragmentation first appeared at 9 mmol/L after 8 h and formed at all concentrations after 20 h (Figure 5). It is well known that cytochrome c released from mitochondria into the cytosol triggers the activation of caspase 9 and caspase 3 in the mitochondrial pathway. In view of this, we propose the involvement of the mitochondrial pathway via cytochrome c release in CCl4-induced apoptosis.
In the present study, we investigted the level of Bcl-XL, an anti-apoptotic protein, in CCl4-induced apoptosis in rat primary hepatocytes. Bcl-XL, the only member of the Bcl-2 family present in hepatocytes, is one of the markers used to identify apoptosis in rat primary hepatocytes[18]. Bcl-XL exerts an anti-apoptosis function by interacting with cytochrome c, either directly or indirectly via Apaf-1, which binds to both Bcl-XL and cytochrome c[2,4,19,20]. Therefore, decreased Bcl-XL levels and increased cytochrome c release are specific biomarkers for the mitochondrial pathway of apoptosis[18]. In our experiments, cytochrome c levels increased markedly after a 8-h CCl4 treatment with a concomitant decrease in Bcl-XL protein expression. Bcl-XL may not execute its anti-apoptotic function once large amounts of cytochrome c are released from the mitochondria when the cell is over-exposed to stimuli, for example, lipid peroxidation induced by CCl4 in this system. These findings strongly suggest the involvement of cytochrome c release from mitochondria accompanied by Bcl-XL regulation in CCl4-induced apoptosis in rat primary hepatocytes.
Hepatic injury through CCl4-induced lipid peroxidation is used extensively in experimental models to elucidate the cellular mechanisms behind oxidative damage[11,21–23]. In the present study, we confirm time- and dose-dependent depletion in the intracellular GSH content after CCl4 treatment. MDA, an important lipid peroxidation product, increased in a time- and dose-dependent manner in our system. The data suggest that oxidative damage is one of the essential mechanisms of hepatotoxicity induced by CCl4 in vitro.
The relationship between ROS and apoptosis has been under investigation for a number of years[4,9]. ROS play important roles in apoptosis initiated in mitochondria[24,25]. It has been documented that GSH depletion and MDA increase, events observed frequently during oxidative damage, are inducers of mitochondrial permeability transition (MPT)[11,26]. Triggering of MPT leads to the release of several different factors relevant to apoptosis, such as cytochrome c, Apoptosis-inducing factor (AIF), and endonuclease G[2,8,9,27]. In this study, we demonstrated GSH depletion, MDA increase and apoptosis initiated from mitochondria of CCl4-treated rat primary hepatocytes. Our results collectively indicate that CCl4 induces apoptosis partly via a mitochondria-dependent pathway, dependent on ROS production in rat primary hepatocytes.
To elucidate other possible mechanisms of CCl4-induced apoptosis, we analyzed the expression of endonuclease G, a caspase-independent apoptotic protein released from mitochondria that translocates to the nucleus during apoptosis[28,29]. Western blot analysis of endonuclease G revealed no evident release in rat primary hepatocytes treated with CCl4 (data not shown). The finding implies that endonuclease G does not participate in apoptosis induced by CCl4.
In summary, our results confirm that oxidative damage is one of the essential mechanisms of hepatotoxicity induced by CCl4. Moreover, mitochondria-initiated apoptosis triggered by ROS plays an important role in this hepatotoxicity in rat primary hepatocytes.
References
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002;192:131-7.
- Van Gurp M, Festjens N, Van Loo G, Saelens X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 2003;304:487-97.
- Zhang J, Dong M, Li L, Fan Y, Pathre P, Dong J, et al. Endonuclease G is required for early embryogenesis and normal apoptosis in mice. Proc Natl Acad Sci USA 2003;100:15782-7.
- Susin SA, Zamzami N, Kroemer G. Mitochondria as regulators of apoptosis: doubt no more. Biochim Biophys Acta 1998;1366:151-65.
- Lorenzo HK, Susin SA. Mitochondrial effectors in caspase-independent cell death. FEBS Lett 2004;557:14-20.
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001;15:2922-33.
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479-89.
- Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta 1998;1366:127-37.
- Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 1998; 273: 11 401–4.
- Sun F, Hamagawa E, Tsutsui C, Ono Y, Ogiri Y, Kojo S. Evaluation of oxidative stress during apoptosis and necrosis caused by carbon tetrachloride in rat liver. Biochim Biophys Acta 2001;1535:186-91.
- Beddowes EJ, Faux SP, Chipman JK. Chloroform, carbon tetrachloride and glutathione depletion induce secondary genotoxicity in liver cells via oxidative stress. Toxicology 2003;187:101-15.
- Shi J, Aisaki K, Ikawa Y, Wake K. Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am J Pathol 1998;153:515-25.
- Berry MN, Friend DS. High-yield preparation of isolated parenchymal cells: a biochemical and fine structural study. J Cell Biol 1969;43:506-20.
- Orrenius S, Thor H, Rajs J, Berggren M. Isolated rat hepatocytes as an experimental tool in the study of cell injury. Effect of anoxia. Forensic Sci 1976;8:255-63.
- Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol 1976;13:29-83.
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 1988;15:61-9.
- Araragi S, Kondoh M, Kawase M, Saito S, Higashimoto M, Sato M. Mercuric chloride induces apoptosis via a mitochondrial-dependent pathway in human leukemia cells. Toxicology 2003;184:1-9.
- Gómez-Lechón MJ, O’Connor E, Castell JV, Jover R. Sensitive markers used to identify compounds that trigger apoptosis in cultured hepatocytes. Toxicol Sci 2002;65:299-308.
- Skulachev VP. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett 1998;423:275-80.
- Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-XL. J Biol Chem 1999;274:2225-33.
- Recknagel RO, Glende EA Jr, Dolak JA, Waller RL. Mechanisms of carbon tetrachloride toxicity. Pharmacol Ther 1989;43:139-45.
- Zimmerman HJ. Hepatotoxicity: The adverse effects of drugs and other chemicals on the liver. 2nd ed. Indirect cytotoxic hepatotoxins. New York: Appleton-Century-Crofts; 1978. p 255–95.
- Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology 2003;189:113-27.
- Pereira CF, Oliveira CR. Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis. Neurosci Res 2000;37:227-36.
- Ding WX, Nam Ong C. Role of oxidative stress and mitochondrial changes in cyanobacteria-induced apoptosis and hepatotoxicity. FEMS Microbiol Lett 2003;220:1-7.
- Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, et al. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J 2001;15:741-51.
- Hengartner MO. Apoptosis. DNA destroyers. Nature 2001;412:27-9.
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001;412:95-9.
- Van Loo G, Schotte P, Van Gurp M, Demol H, Hoorelbeke B, Gevaert K, et al. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Diff 2001;8:1136-42.