
Neuroprotective effects of cyclooxygenase-2 inhibitor celecoxib against toxicity of LPS-stimulated macrophages toward motor neurons
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive fatal neurodegenerative disorder that primarily affects motor neurons in the cortex, brainstem and spinal cord. Evidence suggests that mutations in Cu/Zn superoxide dismutase (SOD-1), glutamate-mediated excitotoxicity, free radical-mediated damage, mitochondrial dysfunction and apoptosis may be involved in the pathogenesis of ALS[1]. However, the cause of ALS is not completely understood.
Accumulating evidence indicates that inflammatory processes, especially the activation of microglia, are involved in the pathogenesis of ALS[2]. Activated microglia are present before the onset of clinical symptoms and prior to significant motor neuron loss in transgenic mice with mutations of the SOD-1gene, an animal model of ALS[3]. Furthermore, some critical markers of microglia activation have also been found post-mortem in the cerebral cortex and spinal cord of patients with ALS[4,5]. Inflammatory processes would produce harmful effects on neuron survival in ALS tissues, include a prominent upregulation of inducible nitric oxide synthase (iNOS) activity with the subsequent generation of nitric oxide (NO)[6], the increased generation of reactive oxygen species (ROS) and glutamate[7], the enhanced secretions of inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6)[8], as well as the enhanced expression of cyclooxygenase-2 (COX-2) with the subsequent production of prostaglandin E2 (PGE2)[9].
COX-2 was demonstrated to be crucial for prostaglandin synthesis in inflammation. COX-2 expression was shown to be dramatically increased in the spinal cord of both of ALS transgenic mice and ALS patients[10,11]. PGE2 levels were also markedly increased in ALS cases compared to non-ALS specimens[12]. In addition, a selective COX-2 inhibitor, SC236, protected motor neurons in an organotypic cell culture model of ALS[13]. Furthermore, celecoxib, a highly selective COX-2 inhibitor clinically available for the treatment of rheumatoid arthritis[14], was proved to prolong survival in a transgenic mouse model of ALS[15]. These results support a potential role for COX-2 in the neurodegenerative processes of ALS and suggest that a selective COX-2 inhibitor may be effective in the treatment of ALS. However, research on the neuroprotective mechanism of COX-2 inhibition on a cellular level, and drug screens using an injured motor neuronal model are still lacking.
In order to investigate the possible neuroprotective mechanism of the COX-2 inhibitor on ALS and to screen candidate anti-inflammatory drugs for ALS, an injured motor neuronal model, which simulates in vivo human microglia activation and the neuronal damage observed during neurodegenerative disease processes, was developed in the present study. Microglia are the resident macrophages of the central nervous system (CNS) as microglia and macrophages, both being cells of the monocyte phagocytic system, have similar biochemical characteristics[16]. In addition, recent findings suggest that infectious agents may increase the risk of ALS and infected migratory mononuclear phagocytes may play an important role in the infection process[17]. Therefore mouse peritoneal macrophages as an accessible source of mononuclear phagocytes and neurotoxicity were used. NSC34 cells, a hybrid cell line obtained by fusion of motor neuron-enriched embryonic mouse spinal cord cells with mouse neuroblastoma N18TG cells, were used as the target motor neuronal cells[18]. Lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria, can activate macrophages. Interferon (IFN)-γ is another important stimulant which can enhance cytokine production. A combination of LPS and IFN-γ was used to activate monocytes[19,20].
In the present study, we further verified the injured motor neuronal model by evaluating the neuroprotective effect of celecoxib, which prolongs the survival of ALS transgenic mice. In addition, the release of PGE2, NO, ROS, inflammatory cytokines and the expression of relevant inflammatory genes in macrophages was studied to explore the possible mechanism of the neuroprotective effect of a COX-2 inhibitor against the toxicity of microglia activation on motor neuron viability.
Materials and methods
Drugs and reagents Celecoxib was kindly provided by Dr Yu-she YANG, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China. LPS (Escherichia coli 055:B5) was purchased from Sigma (St Louis, USA). IFN-γ was obtained from Clonbiotech (Shanghai, China). A radioimmunoassay (RIA) kit for PGE2 was obtained from China PLA General Hospital (Beijing, China). Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α and IL-1β were obtained from BD Biosciences (San Diego, CA, USA).
Cell culture Macrophages were obtained from the peritoneal exudates of female BALB/c mice (Grade II, Shanghai Experimental Animal Center, Shanghai, China; Certificate N
The mouse motor neuronal NSC-34 cell line was a gift from Dr Jin REN (Shanghai Institute of Materia Medica). NSC34 cells were maintained at 37 °C and 5% CO2 in DMEM (Gibco) supplemented with 10% FCS, 0.1 g/L streptomycin and 100 kU/L penicillin.
Measurement of cell viability NSC34 cell viability was measured using the MTT assay as described by Hansen et al[21]. Briefly, MTT (Sigma) was added to cell cultures to reach a final concentration of 0.2 g/L. Following a 4-h incubation at 37 °C, the dark crystals formed were collected and dissolved in 200 µL/well dimethylsulfoxide in 24-well plates. Subsequently, optical densities were measured at 570 nm by transferring 100 µL/well to 96-well plates and recording the values using a plate reader (POLARstar®; BMG, Victoria, Australia). The number of viable NSC34 cells was calculated as a percentage of the value obtained from the control NSC34 cells incubated with media only.
Measurement of nitric oxide Nitric oxide was determined by assaying for nitrite using Greiss reagent (1% sulfanila-mide, 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 5% H3PO4)[22]. Briefly, 100 µL of the supernatant from each well (24-well plates) was incubated for 5 min with 100 µL Griess reagent in 96-well plates. Optical densities of the samples were then obtained by reading absorbance at 540 nm.
Measurement of reactive oxygen species The ROS assay was modified from the fluorescence assay described by Gunasekar et al[23]. Briefly, 90 µL of supernatant from each well (24-well plates) was transferred to 96-well fluorescence assay plates, incubated to 37 °C, and then added 10 µL Krab's Ringer buffer (127 mmol/L NaCl, 5.5 mmol/L KCl, 2 mmol/L MgSO4, 1 mmol/L CaCl2, 20 mmol/L HEPES, 10 mmol/L dextrose, pH 7.4) with 50 µmol/L 2',7'-dichlorofluorescein diacetate (DCFH-DA; Sigma) and 20 IU/mL horseradish peroxide(SABC, Shanghai, China). The fluorescence value of each well was read at 30 s intervals for 5 min at 485 nm excitation wavelength and 520 nm emission wavelength. The ROS value of each sample was calculated as the slope of its time-fluorescence value curve.
Measurement of PGE2, TNF-α and IL-1β The concentrations of PGE2, TNF-α and IL-1β were determined by RIA and ELISA according to the manufacturer's instructions.
Measurement of mRNA The mRNA levels of COX-2, iNOS, TNF-α and IL-1β were detected by real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) using β-actin mRNA as an internal control. Briefly, macrophages from various treatments in 6-well plates were washed twice with ice-cold PBS. Total RNA was extracted from the cells in each well with RNAzol (Dingguo Biotechnology, Beijing, China) according to the manufacturer's instructions. First-strand cDNA synthesis was carried out using 2 µg total RNA and M-MLV reverse transcriptase (Promega, Madison, USA). The reaction mix was incubated at 42 °C for 60 min, and then heated at 70 °C for 10 min. Each reaction mixture was diluted 4 times with 0.1% diethyl pyrocarbonate (DEPC)-treated H2O. A 2-µL aliquot from each diluted reaction mixture was used for real-time PCR amplification. The primer sequences and PCR cycle parameters for β-actin[24], COX-2[24], iNOS[25], TNF-α[26],and IL-1β[27] are listed in Table 1.
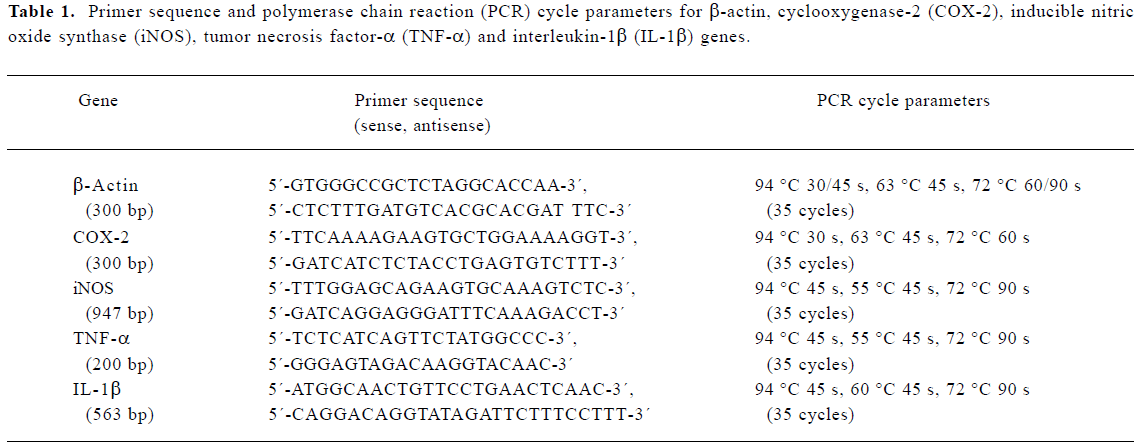
Full table
Real-time quantitative RT-PCR was carried out as described by Livak[28]. Each reaction contained 2 µL of the cDNA sample with 1 IU Taq DNA polymerase (Dingguo Biotechnology) and 0.5 µL Sybr Green (OPE, Shanghai, China) in a total volume of 20 µL in a real-time quantitative PCR cycler (DNA Engine Opticon? Continuous Fluorescence Detection System; MJ Research, USA). The mRNA level was estimated as a relative value by normalizing with β-actin mRNA. Reaction products were also separated on a 1.5% agarose gel, stained with ethidium bromide, and photographed to validate the reliability of the objective genes.
Statistical analysis Data were presented as mean±SD of the values from 3 independent experiments and the Student's t-test was used for the comparison. Values of P<0.05 were considered statistically significant.
Results
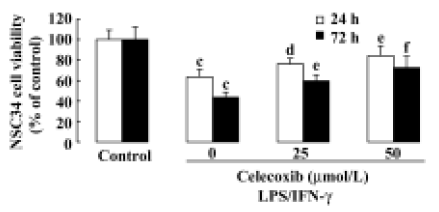
Toxicity of LPS+IFN-γ-stimulated macrophages toward NSC34 cells and inhibitory effect of celecoxib on cytotoxicity The supernatant of the macrophages stimulated with a combination of LPS and IFN-γ significantly inhibited the viability of motor neuron NSC34 cells (Figure 1). Adding celecoxib enhanced the survival of these NSC34 cells (Figure 1). The neuroprotective effect of celecoxib was due to an effect on macrophage neurotoxic secretions because celecoxib showed no neuroprotective effect on NSC34 cells if it was added to the supernatant of NSC34 cells directly (data not shown).
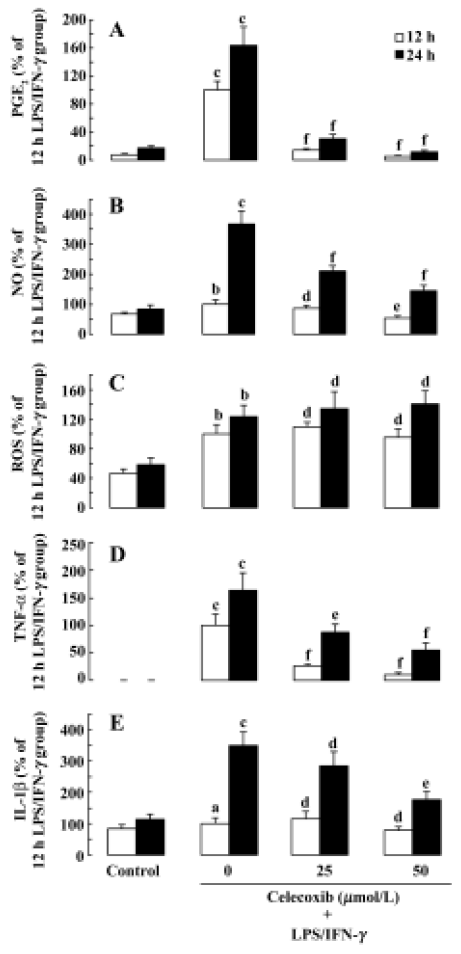
Effect of celecoxib on the production of PGE2, NO, ROS, TNF-α and IL-1β on LPS+IFN-γ-stimulated macrophages To confirm that the production of PGE2, NO, ROS, TNF-α and IL-1β was increased in LPS+IFN-γ-stimulated macrophages, their concentrations were measured in the macrophage supernatants. Compared with resting macrophages, LPS+IFN-γ-stimulated macrophages significantly increased the release of PGE2, NO, ROS, TNF-α, and IL-1β (Figure 2A–2E). To determine whether celecoxib regulates PGE2, NO, ROS, TNF-α, and IL-1β release from activated macrophages, macrophages were preincubated with celecoxib for 30 min prior to the addition of LPS+ IFN-γ. After the macrophages were stimulated for 12 h or 24 h, the concentrations of PGE2, NO, ROS, TNF-α, and IL-1β in the macrphage supernatants were measured. The increased productions of PGE2, NO, TNF-α, and IL-1β were attenuated by preincubation with celecoxib (Figure 2A–2E). However, pretreatment with celecoxib had no effect on the level of ROS in macrophage supernatants (Figure 2C).
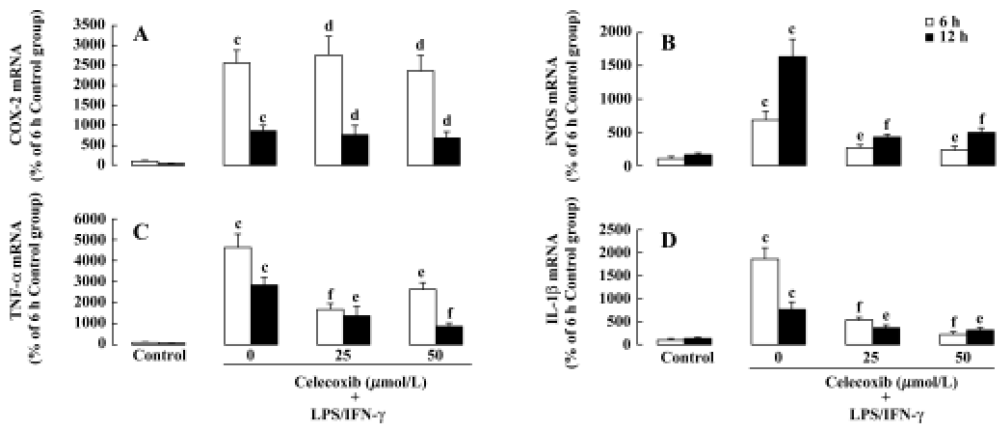
Effect of celecoxib on mRNA levels of COX-2, iNOS, TNF-α and IL-1β in LPS+IFN-γ-stimulated macrophages To determine whether LPS stimulation increases the mRNA levels of COX-2, iNOS, TNF-α, and IL-1β in macrophages, the mRNA levels of these genes were determined by real time RT-PCR. Stimulated macrophages strongly expressed mRNA of COX-2, iNOS, TNF-α and IL-1β following treatment with LPS+IFN-γ (Figure 3A,3D), whereas resting macrophages expressed these mRNA very weakly. Furthermore, preincubation with celecoxib inhibited the increases of the mRNA levels of iNOS, TNF-α and IL-1β, which were induced by LPS+IFN-γ in macrophages and had no effect on the mRNA level of COX-2 (Figure 3).
Discussion
In the present study, we demonstrated that the supernatant of macrophages stimulated with LPS 0.5 mg/L plus IFN-γ 1.5×105 IU/L caused a loss of NSC34 cells following either a 24-h or 72-h exposure. In consisten with the previous results[19,20,29], no neurotoxicity was observed when the supernatant from unstimulated macrophages was transferred to NSC34 cells, and LPS (0.0005−5.0 mg/L) increased the neurotoxity of the supernatant of macrophages on NSC34 cells dose-dependently. LPS did not show toxic effects on NSC34 cells when it was added to the supernatant of NSC34 cells directly (data not shown). These results indicate that NSC34 cells injured by the supernatant of LPS-stimulated macrophages can be used to develop an injured motor neuronal model, whose neurotoxicity was due mainly to inflammatory secretions of macrophages. In the present invastigation, Celecoxib was used to verify the model because of its beneficial effects on ALS transgenic mice. It was shown that celecoxib significantly enhanced the survival of NSC34 cells. These results indicate that this model may have valuable applications for drug screening and further research the mechanisms involved. No improvement in NSC34 cell viability was observed when celecoxib was added directly to NSC34 cells (data not shown). This suggests that the target cells of celecoxib are macrophages rather than motor neurons and that the action of celecoxib on macrophages is likely a reduction of neurotoxic macrophage secretions. Another COX-2 selective inhibitor, NS398, has been reported to have neuroprotective effects on neuronal-like SH-SY5Y cells by suppressing the toxic actions of human monocytic THP-1 cells[20,29].
PGE2 is an important mediator involved in a variety of inflammatory processes and COX-2 has been shown to be primarily responsible for the synthesis of PGE2. COX-2 is rapidly induced by various proinflammatory agents, including LPS, cytokines and mitogens[30]. In the present study, exposure of LPS to macrophages resulted in an increase in the level of PGE2 released and an induction of COX-2 expression at the mRNA level (Figure 3). Results from previous studies have also shown that proinflammatory stimuli induce PGE2 release and COX-2 expression[30]. Our results showed that PGE2 release was inhibited by the addition of celecoxib in LPS-stimulated macrophages (Figure 2), suggesting that celecoxib exerts neuroprotective effects on motor neuron NSC34 cells by inhibiting COX-2 activity and the subsequent production of PGE2 in LPS-stimulated macrophages.
The expression and activity of iNOS plays a pivotal role in sustained and elevated NO release[31]. It has previously been reported that microglial cells are the main source of LPS-induced iNOS/NO both in neuron-glial culture and in vivo[32]. In addition, there is “ross-talk” between iNOS/NO and COX-2/PGE2[33]. In the present study, we demonstrated that celecoxib significantly inhibits LPS-stimulated iNOS expression and NO release in macrophages. These results suggest that downregulation of iNOS/NO by celecoxib might be involved in the neuroprotective effect of celecoxib against LPS-induced motor neuronal death[32].
Although one previous study found that LPS-induced ROS was increased in neurons, not microglial cells, in neuron-glial coculture[33], the most abundant source of oxygen free radicals in the CNS is the respiratory burst system of activated microglia[2]. A recent study reported that LPS treatment increased intracellular ROS in rat microglia in a dose-dependent manner and ROS played a regulatory role in the expression of COX-2 and the subsequent production of PGE2 during the process of microglial activation[34]. On the other hand, Gunasekar et al reported that pretreatment with NS398 significantly decreased potassium cyanide (KCN)-induced ROS generation in cerebellar granule cells. The results indicated the involvement of COX-2 in KCN-induced oxidant generation[23], which further suggests a level of “cross-talk” between ROS and COX-2 in activated microglia. The present studies showed that extracellular ROS levels from LPS-stimulated macrophages were upregulated. However, celecoxib showed no effect on the extracellular ROS level in our study. The results suggest that celecoxib exerts its neuroprotective effect against the toxicity of LPS-stimulated macrophages probably not by the regulation of extracellular ROS.
TNF-α is a potent proinflammatory cytokine that plays an important role in immunity and inflammation. The present study showed that LPS-stimulation increases TNF-α secretion from macrophages. A previous study found that LPS-stimulation increases TNF-α secretion in microgila such as BV-2 cells[19]. We further demonstrated that celecoxib significantly downregulates TNF-α mRNA level and TNF-α secretion induced by LPS in macrophages. The results suggest that inhibition of TNF-α secretion from the LPS-activated macrophages probably participates in the neuroprotective effect of celecoxib on motor neurons.
Interleukin-1β is an important cytokine in the inflammation process, and microglia are an important source of IL-1 in the human CNS[35]. The release of IL-1 plays a critical role in the effect of microglial activation on motor neuron viability and IL-1 is amongst a wide range of factors that upregulate the expression of COX-2 and the subsequent production of proinflammatory cytokines and PGE2[9]. Previous studies reported that LPS increases the secretions of IL-1β in monocytic THP-1 cells[35]. In addition, another study indicated that celecoxib decreases the secretion of IL-1β in rats[36]. The data from the present study show that celecoxib inhibits the level of IL-1β, which is increased in LPS-stimulated macrophages. Therefore, the inhibitory effect of celecoxib on the level of IL-1β may be involved in the mechanism of its neuroprotective effect.
In summary, an injured motor neuronal model was established. The selective COX-2 inhibitor celecoxib showed beneficial effects against motor neuronal death induced by inflammatory reaction. The neuroprotective effect of celecoxib might be associated with downregulation of the levels of PGE2, NO, TNF-α and IL-1β as well as gene expression of iNOS, TNF-α and IL-1β. Since PGE2[12], NO[37-39] and TNF-α[19,40] have been reported to be upregulated in ALS, indicating COX-2 inhibitors would be promising candidates for the treatment of ALS.
References
- Dib M. Amyotrophic lateral sclerosis progress and prospects for treatment. Drugs 2003;63:289-310.
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002;26:459-70.
- Alexianu ME, Kozovska ME, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001;57:1282-9.
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 1992;140:691-707.
- Troost D, Van den Oord JJ, Vianney de Jong JM. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 1990;16:401-10.
- Abe K, Abe Y, Saito H. Agmatine suppresses nitric oxide production in microglia. Brain Res 2000;872:141-8.
- Piani D, Frei K, Do KQ, Cuenod M, Fontana A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 1991;133:159-62.
- Chao CC, Hu S, Peterson PK. Glia, cytokines, and neurotoxicity. Crit Rev Neurobiol 1995;9:189-205.
- Levi G, Minghetti L, Aloisi F. Regulation of prostanoid synthesis in microglial cells and effects of prostacyclin E2 on microglial functions. Biochimie 1998;80:899-904.
- Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 2001;49:176-85.
- Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL. Marked increase in cyclooxygenase-2 in ALS spinal cord: implications for therapy. Neurology 2001;576:952-6.
- Maihofner C, Probst-Cousin S, Bergmann M, Neuhuber W, Neundorfer B, Heuss D. Expression and localization of cyclooxygenase-1 and -2 in human sporadic amyotrophic lateral sclerosis. Eur J Neurosci 2003;18:1527-34.
- Drachman DB, Rothstein JD. Inhibition of cyclooxygenase-2 protects motor neurons in an organotypic model of amyotrophic lateral sclerosis. Ann Neurol 2000;48:792-5.
- Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase-2 in inflammation and pain. Proc Natl Acad Sci USA 1994;91:12013-7.
- Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, et al. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol 2002;52:771-8.
- Thomas WE. Brain marcrophages: evaluation of microglia and their functions. Brain Res Brain Res Rev 1992;17:61-74.
- Portegies P, Cohen ES. Possible etiological role retroviruses and enteroviruses in the development ofamyotrophic lateral sclerosis. Ned Tijdschr Geneeskd 2002;146:1398-400.
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, et al. Neuroblastoma X spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 1992;194:209-21.
- He BP, Wen WY, Strong MJ. Activated microglia (BV-2) facilitation of TNF-α-mediated motor neuron death in vitro. J Neuroimmunol 2002;128:31-8.
- Klegeris A, McGeer PL. Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol Aging 2002;237:787-94.
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Meth 1989;119:203-10.
- Eigler A, Moeller J, Endres S. Exogenous and endogenous nitric oxide attenuates tumor necrosis factor synthesis in ht murine macrophage cell line RAW 264.7. J Immunol 1995;154:4048-54.
- Gunasekar PG, Borowitz JL, Isom GE. Cyanide-induced generation of oxidative species: involvement of nitric oxide synthase and cyclooxygenase-2. J Pharmacol Exp Ther 1998;285:236-41.
- Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther 2000;293:417-25.
- Jeon HK, Jung NP, Choi IH, Oh YK, Shin HC, Gwag BJ. Substance P augments nitric oxide production and gene expression in murine macrophages. Immunopharmacology 1999;41:219-26.
- Suzuki T, Ogata A, Tashiro K, Nagashima K, Tamura M, Yasui K, et al. Japanese encephalitis virus up-regulates expression of macrophage migration inhibitory factor (MIF) mRNA in the mouse brain. Biochim Biophys Acta 2000;1517:100-6.
- Choi CY, Kim JY, Kim YS, Chung YC, Hahm KS, Jeong HG. Augmentation of macrophage functions by an aqueous extract isolated from Platycodon grandiflorum. Can Lett 2001;166:17-25.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆CT method. Methods 2001;25:402-8.
- Klegeris A, Walker DG, McGeer PL. Toxicity of human THP-1 monocytic cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs (NSAIDS). Neuropharmacology 1999;38:1017-25.
- Callejas NA, Castrillo A, Bosca L, Marin-Sanz P. Inhibition of prostaglandin synthesis up-regulates cyclooxygenase-2 induced by lipopolysaccharide and peroxisomal proliferators. J Pharmacol Exp Ther 1999;288:1235-41.
- Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J 1992;6:3051-64.
- Heneka MT, Feinstein DL. Expression and function of inducible nitric oxide synthase in neurons. J Neuroimmunol 2001;114:8-18.
- Kim EJ, Kwon KJ, Park JY, Lee SH, Moon CH, Baik EJ. Neuroprotective effects of prostaglandin E2 or cAMP against microglia and neuronal free radical mediated toxicity associated with inflammation. J Neurosci Res 2002;70:97-107.
- Wang T, Qin L, Liu B, Liu Y, Wilson B, Eling TE, et al. Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem 2004;88:939-47.
- Klegeris A, McGeer PL. Inflammatory cytokine levels are influenced by interactions between THP-1 monocytic, U-373 MG astrocytic, and SH-SY5Y neuronal cell lines of human origin. Neurosci Lett 2001;31:341-4.
- Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res 2002;68:337-43.
- Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat 1996;10:249.
- Sasaki S, Shibata N, Komori T, Iwata M. iNOS and nitro tyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci Lett 2000;291:44.
- Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase upregulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 1999;72:2415-25.
- Poloni M, Facchetti D, Mai R, Micheli A, Agnoletti L, Francolini G, et al. Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett 2000;287:211-4.